Tariquidar (XR9576)
别名: XR9576
Tariquidar是一种有效的,选择性的,非竞争性P-glycoprotein抑制剂,在CHrB30细胞系中Kd为5.1 nM,作用于MDR细胞系逆转耐药性。Phase 3。

Tariquidar (XR9576) Chemical Structure
CAS: 206873-63-4


产品质控
批次:
纯度:
99.81%
99.81
Tariquidar (XR9576)相关产品
| 相关产品 | Zosuquidar 3HCl Elacridar (GF120918) (20S)-Protopanaxadiol Solamargine Sinapine UCLA GP130 2 MRP2 / ABCC2 Antibody (Rabbit mAb) [A16B6] | 点击展开 |
|---|---|---|
| 相关化合物库 | FDA药物库 天然产物库 离子通道配体库 外泌体分泌相关化合物库 钙通道阻滞剂库 | 点击展开 |
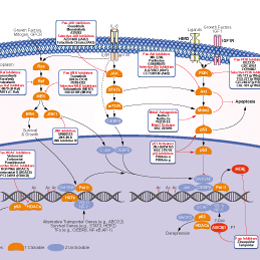
相关信号通路图
细胞实验数据示例
| 细胞系 | 实验类型 | 给药浓度 | 孵育时间 | 活性描述 | 文献信息(PMID) |
|---|---|---|---|---|---|
| SW620 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human SW620 cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | |
| HepG2 | Function assay | 10 uM | 90 mins | Inhibition of P-gp mediated efflux in adriamycin-resistant human HepG2 cells assessed as intracellular rhodamine-123 accumulation at 10 uM incubated in dark condition for 90 mins by flow cytometry relative to control | 27328029 |
| MDCK | Function assay | 30 mins | Activity at BCRP (unknown origin) expressed in MDCK cells using rhodamine 123 as substrate incubated for 30 mins prior to substrate addition measured after 30 mins by fluorometric analysis, EC50=0.01μM | 23374872 | |
| OVCAR8 | Function assay | 1000 nM | 72 hrs | Potentiation of cytotoxicity against human OVCAR8 cells assessed as IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 8.53 +/- 1.95 nM), IC50=0.00518μM | 27504669 |
| K562/A02 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562/A02 cells after 48 hrs by MTT assay, IC50=27.19μM | 28645831 | |
| Kb-V1 | Function assay | 10 mins | Inhibition of ABCB1 expressed in Kb-V1 cells after 10 mins by calcein-AM assay, IC50=0.223μM | 21570282 | |
| KBv1 | Function assay | Inhibition of ABCB1 overexpressed in human KBv1 cells by flow cytometric-based calcein-AM efflux assay, IC50=0.223μM | 19170519 | ||
| KBV1 | Function assay | Inhibition of ABCB1 in human KBV1 cells assessed as inhibition of calcein-AM efflux, IC50=0.22μM | 26774038 | ||
| A2780 | Function assay | 30 mins | Inhibition of human Pgp in A2780 cells after 30 mins by Hoechst 33342 assay, IC50=0.12589μM | 18083034 | |
| KB-3-1 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against human KB-3-1 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.15 +/- 0.04 uM), IC50=0.11μM | 27504669 |
| OVCAR8 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against human OVCAR8 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.12 +/- 0.03 uM), IC50=0.08μM | 27504669 |
| A2780/ADR | Function assay | Inhibition of P-glycoprotein-mediated multidrug resistance in adriamycin-resistant human A2780/ADR cells by calcein AM assay, IC50=0.078μM | 19250834 | ||
| A2780adr | Function assay | Inhibition of P-gp expressed in A2780adr cells by calcein AM accumulation assay, IC50=0.08μM | 21354800 | ||
| A2780 | Function assay | Inhibition of P-gp in human adriamycin-resistant A2780 cells by Hoechst 33342 assay, IC50=0.07244μM | 18678495 | ||
| KBV1 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in KBV1 cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 5.07 +/- 0.19 uM), IC50=0.07μM | 27504669 |
| CEM/VLB500 | Function assay | 3 days | Reversal of P-gp-mediated multidrug resistance to in human CEM/VLB500 cells after 3 days by resazurin assay, EC50=0.068μM | 17399990 | |
| EMT6/AR1.0 | Function assay | 1 hr | Inhibition of mouse Pgp in EMT6/AR1.0 cells after 1 hr by daunorubicin accumulation assay, IC50=0.06457μM | 18083034 | |
| EMT6/AR1.0 | Function assay | 1 hr | Inhibition of mouse Pgp in EMT6/AR1.0 cells after 1 hr by daunorubicin accumulation assay, IC50=0.064μM | 18083034 | |
| MDCK | Function assay | 30 mins | Inhibition of P-glycoprotein (unknown origin) expressed in MDCK cells assessed as reduction of calcein-AM transport after 30 mins by fluorescence assay, EC50=0.044μM | 24607999 | |
| MDCK | Function assay | 30 mins | Activity at MDR1 (unknown origin) expressed in MDCK cells using calcein AM as substrate incubated for 30 mins prior to substrate addition measured after 30 mins by fluorometric analysis, EC50=0.044μM | 23374872 | |
| NCI-ADR-RES | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in NCI-ADR-RES cells assessed as potentiation of cytotoxicity by measuring IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 3714.80 +/- 383.58 nM), IC50=0.01851μM | 27504669 |
| KBV1 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in KBV1 cells assessed as potentiation of cytotoxicity by measuring IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 277.68 +/- 56.61 nM), IC50=0.00066μM | 27504669 |
| KBV1 | Function assay | 10 mins | Inhibition of ABCB1 in human KBV1 cells after 10 mins by Calcein-AM microplate assay, IC50=0.223μM | 24900683 | |
| MCF7/Topo | Function assay | Inhibition of ABCG2 overexpressed in human MCF7/Topo cells by flow cytometric-based mitoxantrone efflux assay, IC50=0.916μM | 19170519 | ||
| MCF7/Topo | Function assay | 2 hrs | Inhibition of ABCG2 in human MCF7/Topo cells after 2 hrs by Hoechst 33342 staining based fluorescence assay, IC50=0.526μM | 30128080 | |
| MCF7/Topo | Function assay | 2 hrs | Inhibition of ABCG2 in human MCF7/Topo cells after 2 hrs by Hoechst 33342 microplate assay, IC50=0.526μM | 24900683 | |
| MCF7/Topo | Function assay | Inhibition of ABCG2 expressed in human MCF7/Topo cells by Hoechst microplate assay, IC50=0.526μM | 21570282 | ||
| MCF7/Topo | Function assay | Inhibition of ABCG2 in human MCF7/Topo cells by Hoechst 33342 assay, IC50=0.52μM | 26774038 | ||
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 measured after 48 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=1.6μM | 28645831 | |
| MCF-7 MX | Function assay | Inhibition of BCRP expressed in MCF-7 MX cells using Hoechst 33342 staining, IC50=1.5μM | 21354800 | ||
| MCF7 | Function assay | Inhibition of ABCG2 in human mitoxantrone-resistant MCF7 cells by Hoechst 33342 assay, IC50=1.44544μM | 18678495 | ||
| HFE | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HFE cells assessed as cell viability after 72 hrs by MTT assay, IC50=1.28μM | 26197160 | |
| MDCK | Function assay | Inhibition of BCRP expressed in MDCK cells using Hoechst 33342 staining, IC50=0.94μM | 21354800 | ||
| KBV | Function assay | 5 uM | 72 hrs | Reversal of P-gp-mediated drug resistance in human KBV cells assessed as potentiation of cytotoxicity by measuring IC50 at 5 uM after 72 hrs by MTT assay (Rvb = 398.34 +/- 0.58 uM), IC50=5.24μM | 30384042 |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 6 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=4.97μM | 28645831 | |
| KBV | Function assay | 10 uM | 72 hrs | Reversal of P-gp-mediated drug resistance in human KBV cells assessed as potentiation of cytotoxicity by measuring IC50 at 10 uM after 72 hrs by MTT assay (Rvb = 398.34 +/- 0.58 uM), IC50=4.46μM | 30384042 |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured immediately by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=3.02μM | 28645831 | |
| K562/A02 | Function assay | 5 uM | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 at 5 uM measured after 48 hrs by MTT assay (Rvb = 43.75 to 96.91 uM), IC50=1.97μM | 28645831 |
| CCRF-CEM/VCR1000 | Function assay | 240 secs | Inhibition of P-glycoprotein-mediated daunorubicin efflux from human CCRF-CEM/VCR1000 cells after 240 secs by FACS flow cytometric analysis, IC50=0.03311μM | 22452412 | |
| MDCK | Function assay | Inhibition of MDR1 expressed in MDCK cells using rhodamine 123 staining by flow cytometry, IC50=0.21μM | 21354800 | ||
| NCI-ADR-RES | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in NCI-ADR-RES cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 5.54 +/- 0.60 uM), IC50=0.24μM | 27504669 |
| MCF7 MX | Function assay | Inhibition of BCRP expressed in MCF7 MX cells by Hoechst 33342 staining, IC50=0.68μM | 19932960 | ||
| MDCK | Function assay | Inhibition of BCRP expressed in MDCK cells by pheophorbide A assay, IC50=0.85μM | 19932960 | ||
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 24 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=14.39μM | 28645831 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 12 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=8.28μM | 28645831 | |
| CCD-18Co | Cytotoxicity assay | 48 hrs | Cytotoxicity against human CCD-18Co cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | |
| SW620/AD300 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human SW620/AD300 cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | |
| HLF | Cytotoxicity assay | 48 hrs | Cytotoxicity against HLF cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=16.69μM | 27328029 | |
| CEM/VLB500 | Growth inhibition assay | 3 days | Growth inhibition of human CEM/VLB500 cells after 3 days by resazurin assay, GI50=13.5μM | 17399990 | |
| A2780adr | Function assay | 10 uM | 30 mins | Inhibition of ABCB1 in human A2780adr cells assessed as increase in accumulation of calcein AM at 10 uM preincubated for 30 mins followed by calcein AM addition measured every 60 secs for 60 mins by fluorescence assay relative to control | 29547272 |
| K562/A02 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562/A02 cells overexpressing P-gp assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=27.19μM | 29631786 | |
| K562 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562 cells after 48 hrs by MTT assay, IC50=31.56μM | 28645831 | |
| SK-N-MC | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-MC cells | 29435139 | ||
| KB-V1 | Function assay | 200 nM | Inhibition of P-gp in human KB-V1 cells assessed as increase in rhodamine 123 accumulation at 200 nM | 21657271 | |
| NB1643 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB1643 cells | 29435139 | ||
| SK-N-SH | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-SH cells | 29435139 | ||
| HEK293 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against HEK293 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by CCK8 assay (Rvb = 5.28 +/- 0.74 nM), IC50=0.00495μM | 27504669 |
| KB-3-1 | Function assay | 1000 nM | 72 hrs | Potentiation of cytotoxicity against human KB-3-1 cells assessed as IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.78 +/- 0.27 nM), IC50=0.00041μM | 27504669 |
| HEK293 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 transfected in HEK293 cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by CCK8 assay (Rvb = 504.65 +/- 44.94 nM), IC50=0.02477μM | 27504669 |
| HCT116 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human HCT116 cells assessed as cell viability after 48 hrs by MTT assay, IC50=12.5μM | 26197160 | |
| MCF7/ADR | Cytotoxicity assay | 48 hrs | Intrinsic cytotoxicity against human MCF7/ADR cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=13.1μM | 27328029 | |
| K562 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562 cells assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=31.56μM | 29631786 | |
| HepG2 | Cytotoxicity assay | 48 hrs | Cytotoxicity against adriamycin-resistant human HepG2 cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=37.2μM | 27328029 | |
| K562/DOX | Function assay | 1 uM | 10 mins | Inhibition of P-gp in human K562/DOX cells assessed as increase in rhodamine-123 efflux in human K562 cells at 1 uM incubated for 10 mins hrs by flow cytometry relative to untreated control | 28113128 |
| 点击查看更多细胞系数据 | |||||
生物活性
| 产品描述 | Tariquidar是一种有效的,选择性的,非竞争性P-glycoprotein抑制剂,在CHrB30细胞系中Kd为5.1 nM,作用于MDR细胞系逆转耐药性。Phase 3。 | ||
|---|---|---|---|
| 靶点 |
|
| 体外研究(In Vitro) | ||||
| 体外研究活性 | Tariquidar高亲和力结合到P-gp,Bmax为275 pmol/mg。Tariquidar与P-gp底物非竞争性地相互作用。Tariquidar作用于CHrB30 细胞,增加这些细胞毒素的稳态积累,达到非P-gp表达的AuxB1细胞中观察的水平,EC50为487 nM。Tariquidar可以抑制 P-gp的对Vanadate敏感的ATPase活性,抑制达 60-70%,有效IC50值为43 nM。Tariquidar高浓度时,可以抑制其他耐药机制。1 μM Tariquidar在体外,可以废除ABCG2(BCRP)介导的Camptothecins耐药性。Tariquidar增强几种药物的细胞毒性,包括Doxorubicin。处理具有内在耐药性的小鼠结肠癌细胞系MC26,Doxorubicin比0.1 μM Tariquidar (36 vs 7 nM)的IC50值低5倍。处理具有获得性化疗抗性的小鼠乳腺癌,人类小细胞肺癌,和和人类卵巢癌细胞系(EMT6/AR1.0, H69/LX4 和 2780 AD),Doxorubicin 比0.1 μM Tariquidar的IC50值低22-150倍。从培养系统除去Tariquidar后,P-gp的抑制作用持续23小时。Tariquidar恢复Doxorubicin作用于MCF7WT乳腺癌细胞系衍生的多细胞肿瘤球体模型的细胞毒性。 |
|||
|---|---|---|---|---|
| 激酶实验 | 稳态药物累积实验 | |||
| 细胞在37°C下5% CO2环境中温育60分钟,达到稳态,反应体积为1 mL。在10-9-10-6 M浓度范围内 调查调节剂XR9576对[3H]-配体累积的影响。DMSO储存液中加入调节剂,最终溶剂浓度为0.2 %(v/v)。细胞收集后,通过液体闪烁计数和归一化细胞蛋白质含量测量累计的药物。 | ||||
| 细胞实验 | 细胞系 | 小鼠乳腺癌细胞系MDR EMT6/AR1.0 | ||
| 浓度 | ~100 nM Tariquidar | |||
| 孵育时间 | 4 天 | |||
| 方法 | 细胞按每孔800个接种在含100 μL 培养基的96孔板中,在37°C下温育4小时。随后加入不同浓度的调节剂或溶剂对照(50μL/孔),再温育1小时,加入细胞毒性药物。加入细胞毒性药物(50μL),每孔按一式四份得到终浓度的范围。再温育4天 ,通过Sulforhodamine B实验测评贴壁细胞的细胞增殖。 |
|||
| 实验图片 | 检测方法 | 检测指标 | 实验图片 | PMID |
| Immunofluorescence | MRP7 |
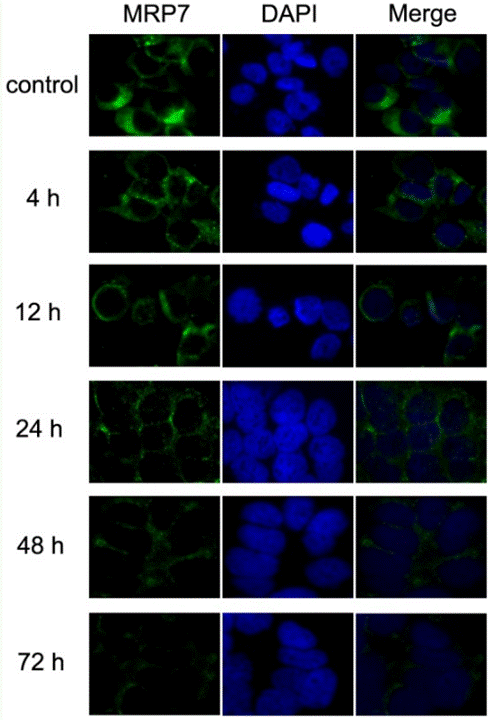
|
23393594 | |
| 体内研究(In Vivo) | ||
| 体内研究活性 | Tariquidar(2-8 mg/kg 口服处理)显著增强Doxorubicin(5 mg/kg, 静脉注射)处理MC26小鼠结肠癌的抗肿瘤活性。Tariquidar和XR9576(6-12 mg/kg 口服处理)合用共处理人类移植瘤, 完全恢复作用于两个高度抗MDR的人类移植瘤(2780AD, H69/LX4)的抗肿瘤活性。 |
|
|---|---|---|
| 动物实验 | Animal Models | 携带结肠癌移植瘤MC26的小鼠 |
| Dosages | 8 mg/kg | |
| Administration | Tariquidar (口服处理)和Doxorubicin (5 mg/kg,静脉注射)共处理 | |
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT01663545 | Completed | Epilepsies Partial |
National Institute of Neurological Disorders and Stroke (NINDS)|National Institutes of Health Clinical Center (CC) |
July 31 2012 | -- |
| NCT01547754 | Terminated | HIV-Associated Cognitive Motor Complex |
National Institute of Mental Health (NIMH)|National Institutes of Health Clinical Center (CC) |
January 9 2012 | -- |
| NCT01386476 | Completed | Drug Resistance |
National Institute of Mental Health (NIMH)|National Institutes of Health Clinical Center (CC) |
June 15 2011 | -- |
| NCT00082368 | Completed | Cancer |
National Cancer Institute (NCI)|National Institutes of Health Clinical Center (CC) |
May 16 2004 | Phase 2 |
|
化学信息&溶解度
| 分子量 | 646.73 | 分子式 | C38H38N4O6 |
| CAS号 | 206873-63-4 | SDF | Download Tariquidar (XR9576) SDF |
| Smiles | COC1=C(C=C2CN(CCC2=C1)CCC3=CC=C(C=C3)NC(=O)C4=CC(=C(C=C4NC(=O)C5=CC6=CC=CC=C6N=C5)OC)OC)OC | ||
| 储存条件(自收到货起) | |||
|
体外溶解度 |
DMSO : 8 mg/mL ( (12.36 mM) ;DMSO吸湿会降低化合物溶解度,请使用新开封DMSO) Water : Insoluble Ethanol : Insoluble |
摩尔浓度计算器 |
|
体内溶解配方 现配现用,请按从左到右的顺序依次添加,澄清后再加入下一溶剂 |
动物体内配方计算器 | |||||
实验计算
动物体内配方计算器(澄清溶液)
第一步:请输入基本实验信息(考虑到实验过程中的损耗,建议多配一只动物的药量)
mg/kg
g
μL
只
第二步:请输入动物体内配方组成(配方适用于不溶于水的药物;不同批次药物配方比例不同,请联系Selleck为您提供正确的澄清溶液配方)
% DMSO
%
% Tween 80
% ddH2O
%DMSO
%
计算结果:
工作液浓度: mg/ml;
DMSO母液配制方法: mg 药物溶于μL DMSO溶液(母液浓度mg/mL,注:如该浓度超过该批次药物DMSO溶解度,请先联系Selleck);
体内配方配制方法:取μL DMSO母液,加入μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入μL ddH2O,混匀澄清。
体内配方配制方法:取μL DMSO母液,加入μL Corn oil,混匀澄清。
注意:1. 首先保证母液是澄清的;
2.一定要按照顺序依次将溶剂加入,进行下一步操作之前必须保证上一步操作得到的是澄清的溶液,可采用涡旋、超声或水浴加热等物理方法助溶。
技术支持
在订购、运输、储存和使用我们的产品的任何阶段,您遇到的任何问题,均可以通过拨打我们的热线电话400-668-6834,或者技术支持邮箱tech@selleck.cn,直接联系到我们。我们会在24小时内尽快联系您。
如果有其他问题,请给我们留言。
* 必填项
常见问题及建议解决方法
问题 1:
Can you please give me more specific and detailed information of how to dissolve and use it (S8028) for in vivo studies?
回答:
It in 30% Propylene glycol, 5% Tween 80, 65% D5W at 30mg/ml will be a suspension or emulsion. If you are going to administrate this compound by oral gavage, it is fine. We also have test some vehicles for it for i.p injection, and it is soluble in 5% DMSO+45% PEG 300+ddH2O at 2mg/ml clearly. When preparing the solution, please dissolve it in DMSO clearly first, then add PEG. After they mixed well, then dilute with water.
