| S1460 |
SP600125
|
SP600125 (Nsc75890)是一种广谱JNK抑制剂,作用于JNK1、JNK2和JNK3,无细胞试验中IC50分别为40 nM、40 nM和90 nM,比作用于MKK4选择性高10倍,比作用于MKK3、MKK6、PKB和PKCα选择性高25倍,比作用于ERK2、p38、Chk1、EGFR等选择性高100倍。SP600125也是一种广谱的serine/threonine kinases抑制剂,包括Aurora kinase A、FLT3和TRKA,对应的IC50值为60 nM、90 nM和70 nM。SP600125可抑制自噬而激活细胞凋亡。 |
-
Research (Wash D C), 2026, 9:1190
-
Anal Chem, 2026, 98(10):7247-7261
-
iScience, 2026, 29(5):115396
|

|
| S7397 |
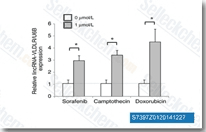
Sorafenib (BAY 43-9006)
|
Sorafenib是Raf-1和B-Raf的多重激酶抑制剂,无细胞试验中IC50分别为6 nM和22 nM。Sorafenib 还可抑制VEGFR-2、VEGFR-3、PDGFR-β、Flt-3和c-KIT,对应的IC50值分别为90 nM、20 nM、57 nM、59 nM和68 nM。Sorafenib 可诱导autophagy、apoptosis并激活ferroptosis,Sorafenib 具有抗肿瘤活性。 |
-
Signal Transduct Target Ther, 2026, 11(1)79
-
Int J Surg, 2026, 10.1097/JS9.0000000000004913
-
Acta Pharmacol Sin, 2026, 10.1038/s41401-026-01791-z
|
|
| S1040 |
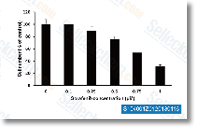
Sorafenib Tosylate (BAY 43-9006)
|
Sorafenib tosylate是Raf-1和B-Raf的多重激酶抑制剂,无细胞试验中IC50分别为6 nM和22 nM。Sorafenib 还可抑制VEGFR-2、VEGFR-3、PDGFR-β、Flt-3和c-KIT,对应的IC50值分别为90 nM、20 nM、57 nM、59 nM和68 nM。Sorafenib Tosylate 可诱导autophagy、apoptosis并激活ferroptosis,并具有抗肿瘤活性。 |
-
Int J Oncol, 2025, 67(3)72
-
Nature, 2024, 629(8013):927-936
-
Cell Mol Life Sci, 2024, 81(1):238
|
|
| S1526 |
Quizartinib (AC220)
|
Quizartinib (AC220)是一种二代FLT3抑制剂,作用于Flt3(ITD/WT),在MV4-11和 RS4;11细胞中IC50分别为1.1 nM/4.2 nM,作用于Flt3比作用于KIT,PDGFRA,PDGFRB,RET,和CSF-1R选择性高10倍。Quizartinib (AC220)可诱导肿瘤细胞的凋亡。Phase 3。 |
-
Cell Rep Med, 2026, 7(3):102687
-
Cell Death Discov, 2026, 12(1):98
-
Signal Transduct Target Ther, 2025, 10(1):222
|

|
| S1048 |
Tozasertib (VX-680, MK-0457)
|
Tozasertib (VX-680)是一种pan-Aurora抑制剂,对Aurora A作用最强,无细胞试验中Kiapp为0.6 nM,而对Aurora B/Aurora C的作用较弱,对Aurora A选择性比其他55种激酶高100倍。唯二的例外是 Fms-related tyrosine kinase-3 (FLT-3) 和 BCR-ABL tyrosine kinase,也可被 Tozasertib 抑制,对应的Ki值均为30 nM和30 nM。Tozasertib 可诱导凋亡和自噬。Phase 2。 |
-
bioRxiv, 2026, 2026.01.11.698900
-
ACS Appl Bio Mater, 2026, 9(8):3753-3767
-
Cell Rep Med, 2025, S2666-3791(25)00102-8
|
|
| S7818 |
Pexidartinib (PLX3397)
|
Pexidartinib (PLX3397)是一种口服有效的多靶点CSF-1R,Kit (c-Kit),和 FLT3受体酪氨酸激酶抑制剂,其IC50分别为20 nM, 10 nM 和 160 nM。Pexidartinib (PLX3397) 可以诱导细胞凋亡和坏死,并具有抗肿瘤活性。Phase 3。 |
-
Nature, 2026, 652(8109):442-450
-
Brain Behav Immun, 2026, 136:106576
-
Oncogene, 2026, 45(21):1970-1987.
|
|
| S2194 |
R406
|
R406(R406 besylate) 是一种有效的 Syk 抑制剂,无细胞试验中IC50为41 nM,对Syk抑制作用强,但是不抑制Lyn,对Flt3的作用比对Syk低5倍。R406 可诱导凋亡。Phase 1。 |
-
Aging (Albany NY), May 7, 2020, 8221-8240
-
eLife, April 30, 2020, e56656
-
Cancer Research, April 15, 2017, 1818-1830
|
|
| S7083 |
Ceritinib (LDK378)
|
Ceritinib是一种有效的ALK抑制剂,在无细胞试验中IC50为0.2 nM。Ceritinib (LDK378)还可抑制IGF-1R、InsR、STK22D和FLT3对应的IC50值分别为8 nM、7 nM、23 nM和60 nM。Phase 3。 |
-
Molecular Cancer Therapeutics, January 01 2018, 175-186
-
Oncotarget, December 29 2015, 44643-44659
-
ESMO Open, August 2021, 100172
|
|
| S1533 |
R406 (free base)
|
R406 (free base) 是一种有效的Syk抑制剂,无细胞试验中IC50为41 nM,强效抑制Syk而不能抑制Lyn,对Flt3作用效果低5倍。R406 (free base) 可触发凋亡。Phase 1。 |
-
Journal of Leukocyte Biology, July 25, 2024, 210-223
-
PLoS One, January 22, 2019, e0210879
-
Blood, February 11, 2016, 739-48
|
|
| S4001 |
Cabozantinib S-malate
|
Cabozantinib malate (XL184)是Cabozantinib的苹果酸盐,是有效的VEGFR2抑制剂,IC50为0.035 nM,也抑制c-Met, RET (c-RET), Kit (c-Kit), Flt-1/3/4, Tie2和AXL,无细胞试验中IC50分别为1.3 nM, 4 nM, 4.6 nM, 12 nM/11.3 nM/6 nM, 14.3 nM和7 nM。Cabozantinib malate (XL184)可诱导细胞凋亡。 |
-
Scientific Reports, 2025, 35889
-
Sci Rep, 2025, 15(1):35889
-
bioRxiv, 2025, 2025.08.15.670608
|
|
| S2736 |
Fedratinib (TG101348)
|
Fedratinib (SAR302503, TG101348)是一种选择性JAK2抑制剂,在无细胞试验中IC50为3 nM,作用于JAK2比作用于JAK1和JAK3选择性高35和334倍。Fedratinib也可抑制 FMS-like tyrosine kinase 3 (FLT3) 和 Ret (c-RET),对应的IC50值分别为15 nM和48 nM。Fedratinib有潜在的抗肿瘤活性。Fedratinib可抑制细胞增殖并促进凋亡。Phase 2。 |
-
Nat Commun, 2026, 17(1):949
-
Leukemia, 2025, 39(7):1678-1691
-
Front Immunol, 2025, 16:1514618
|

|
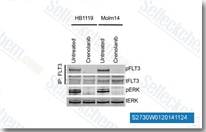
| S2730 |
Crenolanib (CP-868596)
|
Crenolanib是一种有效的,选择性PDGFRα/β抑制剂,在CHO细胞中Kd为2.1 nM/3.2 nM,也能有效抑制FLT3,对D842V突变型敏感对V561D突变型不敏感,作用于PDGFR比作用于c-Kit,VEGFR-2,TIE-2,FGFR-2,EGFR,erbB2,和Src的选择性高100倍以上。Crenolanib可辅助诱导线粒体自噬。 |
-
Exp Mol Pathol, 2026, 146:105037
-
Cancer Cell, 2025, S1535-6108(25)00070-4
-
Nat Commun, 2025, 16(1):1358
|
|
| S1111 |
Foretinib (GSK1363089, XL880)
|
Foretinib是一种ATP竞争性的HGFR和VEGFR抑制剂,对Met (c-Met)和KDR作用最强,在无细胞试验中IC50分别为0.4 nM和0.9 nM。对Ron, Flt-1/3/4, Kit (c-Kit), PDGFRα/β和Tie-2作用效果稍弱,对FGFR1和EGFR几乎没有抑制活性。Phase 2。 |
-
J Microbiol, 2025, 63(2):e2409001
-
Int J Mol Sci, 2023, 24(1)757
-
Biol Open, 2023, 12(8)bio059994
|

|
| S7754 |
ASP2215 (Gilteritinib)
|
Gilteritinib (ASP2215)是小分子FLT3/AXL抑制剂,对FLT3和AXL的IC50值为0.29 nM和0.73 nM。其抑制c-KIT的IC50值约为抑制FLT3的800倍。 |
-
Heliyon, October 12, 2022, e11004
-
bioRxiv, November 24, 2025, nan
-
Cell Death Discov, 2026, 12(1):98
|
|
| S1018 |
Dovitinib (TKI-258)
|
Dovitinib (TKI-258, CHIR-258)是一种多靶点的RTK抑制剂,在无细胞试验中对III型(FLT3/c-Kit)作用最强,IC50为1 nM/2 nM,同时也作用于IV类(FGFR1/3)和V类(VEGFR1-4) RTKs,IC50为8-13 nM,但对InsR,EGFR,c-Met,EphA2,Tie2,IGF-1R和HER2作用较弱。Phase 4。 |
-
Acta Pharmacol Sin, 2026, 10.1038/s41401-026-01791-z
-
Spectrochim Acta A Mol Biomol Spectrosc, 2025, 343:126602
-
Biomed Pharmacother, 2024, 180:117533
|

|
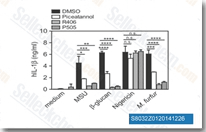
| S8032 |
PRT062607 (P505-15) HCl
|
PRT062607 (P505-15, BIIB057, PRT-2607)是一种新型的,高度选择性Syk抑制剂,无细胞试验中IC50为1 nM,作用于Syk比作用于Fgr,PAK5,Lyn,FAK,Pyk2, FLT3, MLK1 和Zap70选择性高80倍以上。 |
-
Sci Adv, 2025, 11(31):eadu4270
-
Scientific Reports, 2024, 7739
-
Sci Rep, 2024, 14(1):7739
|
|
| S8229 |
Brigatinib
|
Brigatinib是一种有效的、选择性的ALK抑制剂,IC50=0.6 nM;也是ROS1抑制剂,IC50=0.9 nM。它还能以相对较低的效力抑制IGF-1R、FLT3、FLT3(D835Y)和EGFR。 |
-
Leukemia, 2025, 10.1038/s41375-025-02682-8
-
Cell Death Dis, 2025, 16(1):194
-
Toxicol Appl Pharmacol, 2025, 498:117310
|

|
| S2198 |
SGI-1776 free base
|
SGI-1776 free base是一种新型的,ATP竞争性Pim1抑制剂,无细胞试验中IC50为7 nM,比作用于Pim2和Pim3选择性分别高50和10倍,也有效作用于Flt3和haspin。SGI-1776 可诱导凋亡和自噬。 |
-
Cell Death Dis, 2024, 15(9):644
-
Mar Drugs, 2024, 22(10)444
-
Mol Cancer, 2023, 22(1):64
|

|
| S7119 |
Go 6976
|
Go6976 (PD406976)是一种强效的PKC抑制剂,其对PKC (老鼠大脑), PKCα, and PKCβ1的IC50分别为7.9nM,2.3 nM, 和 6.2 nM 。此外,也是JAK2 和 Flt3的强抑制剂。
|
-
FASEB J, 2025, 39(12):e70768
-
bioRxiv, 2025, 2025.02.02.636139
-
Int J Biol Sci, 2024, 20(10):3956-3971
|

|
| S1003 |
Linifanib (ABT-869)
|
Linifanib (ABT-869, AL39324, RG3635)是一种新型有效的ATP竞争性VEGFR/PDGFR抑制剂,作用于KDR,CSF-1R,Flt-1/3和PDGFRβ,其IC50分别为4 nM,3 nM,3 nM/4 nM和66 nM,对突变激酶依赖性癌细胞(即FLT3)最有效。Linifanib (ABT-869) 可诱导自噬和凋亡。Phase 3。 |
-
Acta Pharmacol Sin, 2026, 10.1038/s41401-026-01791-z
-
Research in Pharmaceutical Sciences, 2025 Oct 20, 734-745
-
Biomed Pharmacother, 2024, 180:117533
|
|
| S7765 |
Dovitinib (TKI258) Lactate monohydrate
|
Dovitinib (TKI258) Lactate monohydrate是Dovitinib的乳酸盐,Dovitinib是一种多靶点的PTK抑制剂,主要对第III类(FLT3/c-Kit)发挥作用,IC50为1 nM/2 nM,对第IV 类(FGFR1/3)和第V 类(VEGFR1-4) RTKs也有效,IC50为8-13 nM,对InsR,EGFR,c-Met,EphA2,Tie2,IGFR1 和HER2作用较差。Phase 4。
|
-
Mol Cell, 2021, S1097-2765(21)00507-4
-
Acta Neuropathol, 2021, 10.1007/s00401-021-02327-x
-
Cancer Res, 2021, canres.1780.2020
|
|
| S8057 |
Pacritinib (SB1518)
|
Pacritinib是有效的选择性Janus Kinase 2 (JAK2)和Fms-Like Tyrosine Kinase-3 (FLT3)抑制剂,无细胞试验中IC50分别为23和22 nM。Phase 3。 |
-
Nat Commun, 2026, 17(1):949
-
Front Pharmacol, 2025, 16:1549183
-
Toxicol In Vitro, 2025, S0887-2333(25)00050-5
|

|
| S1244 |
Amuvatinib (MP-470)
|
Amuvatinib (MP-470, HPK 56) 是一种有效的,作用于c-Kit、PDGFα和Flt3的多靶点抑制剂,IC50分别为10 nM、40 nM和81 nM。Amuvatinib 可抑制c-MET和c-RET。Amuvatinib 还具有DNA修复蛋白Rad51抑制剂及抗肿瘤的活性。Phase 2。 |
-
Cancer Res, 2025, 10.1158/0008-5472.CAN-24-2220
-
Hematol Oncol, 2025, 43(5):e70131
-
bioRxiv, 2024, 2023.11.21.568071
|

|
| S2692 |
TG101209
|
TG101209是一种选择性的JAK2抑制剂,无细胞试验中IC50为6 nM,对Flt3和RET (c-RET)作用效果稍弱,IC50分别为25 nM和17 nM,作用于JAK2比作用于JAK3选择性高30倍左右,对JAK2V617F和MPLW515L/K突变型敏感。 |
-
Signal Transduct Target Ther, 2025, 10(1):131
-
Phytomedicine, 2023, 117:154918
-
EBioMedicine, 2022, 78:103963
|
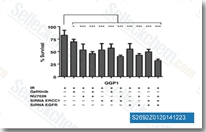
|
| S1043 |
Tandutinib (MLN518)
|
Tandutinib (MLN518, CT53518, NSC726292)是一种有效的FLT3拮抗剂,IC50为0.22 μM,同时也抑制PDGFR和c-Kit,对FLT3作用效果比CSF-1R强15到20倍,比FGFR, EGFR和KDR等强100倍以上。Phase 2。 |
-
Cancer Cell, 2021, S1535-6108(21)00659-0
-
Drug Metab Dispos, 2021, 49(1):53-61
-
Genome Med, 2020, 18;12(1):17
|

|
| S1181 |
ENMD-2076
|
ENMD-2076 选择性地作用于 Aurora A 和 Flt3,IC50分别为14 nM和1.86 nM,作用于Aurora A比作用于Aurora B选择性高25倍,对RET、SRC、NTRK1/TRKA、CSF1R/FMS、VEGFR2/KDR、FGFR和PDGFRα作用效果稍弱。ENMD-2076 可抑制各种人类实体瘤和造血癌细胞系的生长,其IC50值为0.025至0.7μM,可诱导凋亡和G2/M期停滞。Phase 2。 |
-
Int J Biol Macromol, 2025, 292:139119
-
Cell, 2021, 184(2):334-351.e20
-
Cancer Discovery, 2021, 2544-2563
|

|
| S2158 |
KW-2449
|
KW-2449是一种多靶点抑制剂,最有效作用于Flt3,IC50为6.6 nM,作用于Flt3, Bcr-Abl和Aurora A适度有效;对PDGFRβ, IGF-1R, EGFR没有抑制效果。Phase 1。 |
-
Molecular Cancer Research, October 2018, 1556-1567
-
Cancer Research, June 6, 2022, 2141-2155
-
Stem Cell Research & Therapy, December 27, 2022, 534
|

|
| S7576 |
UNC2025 HCl
|
UNC2025 HCl 是一种有效且口服生物可利用的双重MER/FLT3抑制剂,IC50分别为0.74 nM 和 0.8 nM,选择性比作用于Axl和 Tyro3大约高20倍。
|
-
bioRxiv, 2025, 2025.07.18.665641
-
Immunity, 2023, 56(8):1778-1793.e10
-
International Journal of Molecular Sciences, 2023, 15903
|
|
| S7014 |
Merestinib (LY2801653)
|
Merestinib (LY2801653) 是一种2型ATP竞争型的慢抑制剂,抑制Met (c-Met)酪氨酸激酶,Ki值为2 nM。药效的停留时间为0.00132 min(-1),t1/2为525 min。Merestinib (LY2801653) 还可抑制MST1R、AXL、ROS1、MKNK1/2、FLT3、MERTK、DDR1和DDR2,其对应的IC50值分别为11 nM、2 nM、23 nM、7 nM、7 nM、10 nM、0.1 nM 和 7 nM。 |
-
Mol Oncol, 2025, 19(8):2366-2387
-
Molecular Oncology, 2025, 2366-2387
-
NPJ Breast Cancer, 2024, 10(1):65
|
|
| S8442 |
TAK-659 Hydrochloride
|
TAK-659 Hydrochloride是一种有效的、选择性的SYK抑制剂。IC50为3.2 nM。对其他激酶具有选择性,对FLT3也能有效抑制,IC50为4.6 nM。 |
-
ACS Pharmacology & Translational Science, May 14, 2025, 615160
-
Experimental and Therapeutic Medicine, April 4, 2025, 110
-
Metabolomics, 2026, 22(3)51
|
|
| S9662 |
UNC2025
|
UNC2025 是一种有效的、口服活性的 FLT3 和 MER 的双重抑制剂,对应的IC50值分别为0.35 nM和0.46 nM。UNC2025 还抑制 AXL、TRKA、TRKC、QIK、TYRO3、SLK、NuaK1、Kit (c-Kit) 和 Met (c-Met),对应的IC50值分别为1.65 nM、1.67 nM、4.38 nM、5.75 nM、5.83 nM、6.14 nM、7.97 nM、8.18 nM 和 364 nM。 |
-
iScience, 2024, 27(7):110226
-
Commun Biol, 2023, 6(1):916
-
Int J Mol Sci, 2023, 10.3390/ijms242115903
|
|
| S7003 |
AZD2932
|
AZD2932 是一种有效的多靶点蛋白酪氨酸激酶抑制剂,对VEGFR-2,PDGFRβ,Flt-3,和 c-Kit 的 IC50 分别为 8 nM,4 nM,7 nM,和 9 nM。
|
-
Cancer Research, 2021, 747-762
-
Cancer Res, 2020, canres.1992.2020
-
Molecules, 2020, 8;25(9) pii: E2220
|
|
| S0278 |
SU5614
|
SU5614 (Chloro-SU5416, Chloro-Semaxanib) 是一种小分子 receptor tyrosine kinases (RTK) 的抑制剂,对 VEGFR-2、c-kit 以及野生型和突变型 FLT3 均有抑制作用。SU5614 可减少细胞增殖并诱导凋亡。 |
-
Frontiers in Cell and Developmental Biology, January 26, 2022, 797047
-
Advanced Science, August 7, 2021, e2101848
-
Am J Pathol, 2024, S0002-9440(24)00326-2
|
|
| S6662 |
AST-487 (NVP-AST487)
|
AST-487 (NVP-AST487),一种N,N'-二苯基脲,是Flt3的竞争性抑制剂,ki值为0.12 μM。除FLT3以外,AST487还抑制RET,KDR,c-KIT 和 c-ABL 激酶,IC50值低于1 μM。 |
-
Cancer Cell, 2022, S1535-6108(22)00312-9
-
International Journal of Molecular Sciences, 2022, 10819
-
Int J Mol Sci, 2022, 23(18)10819
|
|
| S8023 |
TCS 359
|
TCS 359是一种有效的FLT3抑制剂,IC50为42 nM。 |
-
Nat Commun, 2021, 12(1):2148
-
Blood, 2016, 127(23):2890-90
|
|
| S2018 |
ENMD-2076 L-(+)-Tartaric acid
|
ENMD-2076 L-(+)-Tartaric acid 是ENMD-2076的酒石酸,选择性作用于Aurora A和VEGFR(Flt3),IC50分别为14 nM和1.86 nM,作用于Aurora A比作用于Aurora B选择性高25倍,对VEGFR2/KDR和VEGFR3, FGFR1和FGFR2和PDGFRα作用效果稍弱。Phase 2。 |
-
Veterinary and Comparative Oncology, 2016 Mar, 14(1):13-27
-
International Journal of Oncology, 2015 Jul, 47(1):122-32
|
|
| S8899 |
FF-10101
|
FF-10101是一种新型的不可逆的FLT3抑制剂,对FLT3(WT)和 FLT3(D835Y)的ic50分别为0.20 nM 和 0.16 nM。 |
-
Frontiers in Oncology, August 20, 2021, 686765
-
ACS Pharmacology & Translational Science, 2025, nan
-
Frontiers in Oncology, 2021, 686765
|
|
| S6839 |
MRX-2843
|
MRX-2843 (UNC2371) 是酪氨酸激酶 MERTK 和 FLT3 的口服活性双重抑制剂,对应的IC50值分别为1.3 nM和0.64 nM。 |
-
Cancer Immunology Research, 2025, CIR-24-0335
-
Scientific Reports, 2024, 4125
-
Scientific Reports, 2024, 14:4125
|
|
| S7545 |
G-749
|
G-749是一种新型有效的FLT3抑制剂,对FLT3(WT),FLT3(D835Y),和Mer的IC50分别为0.4 nM,0.6nM 和 1nM,对其它酪氨酸激酶效果较低。
|
-
Scientific Reports, 2024, 4125
-
Front Pharmacol, 2021, 12:730241
|
|
| E2387 |
FLT3-IN-2
|
FLT3-IN-2是一种FLT3抑制剂,IC50低于1 μM。 |
-
Pathogens, 2022, 12(1)53
|
|
| S9779 |
Emavusertib (CA-4948)
|
Emavusertib (CA-4948) 是一种有效的、口服生物可利用的 interleukin-1 receptor-associated kinase 4/FMS-like Tyrosine Kinase 3 (IRAK4/FLT3) 的抑制剂,具有抗肿瘤活性。 |
-
Arthritis Rheumatol, 2021, 10.1002/art.42043
|
|
| S1099 |
SKLB4771 (FLT3-IN-1)
|
SKLB4771是一种有效、选择性的人类受体型酪氨酸蛋白激酶FLT3抑制剂,IC50为10 nM。 |
-
Front Pharmacol, 2022, 13:899775
|
|
| S7533 |
AMG 925
|
AMG 925是一种有效的,且口服具有生物活性的双重FLT3/CDK4抑制剂,IC50分别为1 nM 和 3 nM。
|
-
Molecules, 2020, 8;25(9) pii: E2220
|
|
| S7002 |
Zotiraciclib
|
Zotiraciclib (SB1317; TG02)是一种新型小分子CDK/JAK2/FLT3抑制剂,对CDK1、2和9的IC50分别为9、5和3 nM,抑制FLT3的IC50为19 nM,抑制JAK2的IC50为19 nM。 |
|
|
| E1437 |
PHI-101
|
PHI-101 是一种口服的强效第三代 FLT3 抑制剂,可克服对多种耐药突变的耐药性。它具有研究复发或难治性急性髓系白血病 (AML) 的潜力。 |
|
|
| S0550 |
5'-Fluoroindirubinoxime
|
5'-Fluoroindirubinoxime (5’-FIO, compound 13)是一种靛红素的衍生物,也是一种有效的FMS-样酪氨酸激酶3(FMS-like tyrosine kinase 3,FLT3)抑制剂,IC50为15 nM。 |
|
|
| E4598 |
MEN1703(SEL24,SEL24-B489)
|
MEN1703(SEL24-B489,SEL24, Pim/flt3 kinase inhibitor SEL24) 是一种有效的 I 型口服 PIM 和 FLT3-ITD 双重抑制剂,其对 PIM1 的 Kd 值为 2 nM,对 PIM2 的 Kd 值为 2 nM,对 PIM3 的 Kd 值为 3 nM。MEN1703 在治疗急性髓系白血病方面表现出广泛的治疗潜力。 |
|
|
| S9275 |
Isoguanosine
|
Isoguanosine (Crotonoside)抑制 FLT3 和 HDAC3/6 治疗AML。Isoguanosine 是巴豆(Croton tiglium)种子中天然存在的鸟苷的活性异构体。 |
|
|
| S7640 |
Lestaurtinib
|
Lestaurtinib (CEP-701, KT-5555) 是 indolocarbazole K252 的衍生物,也是星形孢菌素类似物。它是一种口服的受体蛋白酪氨酸激酶 (RPTK) 抑制剂,可抑制 FLT3 和 Trk,IC50 分别为 3 nM 和 < 25 nM。它还能抑制 JAK2,IC50 为 0.9 nM。 |
|
|
| S5986 |
FLT3-IN-3
|
FLT3-IN-3 是 FLT3 的强效抑制剂,对 FLT3 WT 和 FLT3 D835Y 的 IC50 值分别为 13 nM 和 8 nM。FLT3-IN-3 在低纳摩尔浓度下可有效抑制 FLT3-ITD 阳性 MV4-11 和 MOLM-13 细胞系的增殖。 |
|
|
| E4720 |
Flonoltinib
|
Flonoltinib (JAK2/FLT3-IN-1) 是一种强效且选择性的JAK2和FLT3双重抑制剂,体外激酶试验中,对JAK2、JAK2V617F和FLT3的IC50分别为0.8、1.4和15 nM。它还能抑制细胞增殖,并诱导噬血细胞淋巴组织细胞增生症(HLH)患者CD8+T细胞、CTLL2细胞和RAW 264.7细胞的G2/M细胞周期停滞和凋亡。 |
|
|
| S6060 |
HPK1-IN-2
|
HPK1-IN-2 是一种有效的和口服活性的 hematopoietic progenitor kinase-1 (HPK1, a serine/threonine Ste20-related protein kinase)、Lck 和 Flt3 的抑制剂,其IC50值分别为 <0.05 μM、<0.5 μM 和 <0.05 μM。HPK1-IN-2 具有抗肿瘤的活性。 |
|
|
| E2658 |
FN-1501
|
FN-1501是Fms样受体酪氨酸激酶3 (Fms-like receptor tyrosine kinase 3, FLT3) 和细胞周期蛋白依赖性激酶(cyclin-dependent kinase, CDK)的有效抑制剂,对CDK2/cyclin A、CDK4/cyclin D1、CDK6/cyclin D1和FLT3的IC50分别为2.47、0.85、1.96和0.28 nM。 |
|
|
| S9892 |
HM43239
|
HM43239 是一种新型强效小分子 FLT3 抑制剂,不仅选择性抑制 FLT3 野生型、ITD 突变体或 TKD 突变,还选择性抑制 FLT3 ITD/TKD 双突变。 HM43239 对 FLT3 WT、FLT3 ITD 和 FLT3 D835Y 激酶的 IC50 分别为 1.1 nM、1.8 nM 和 1.0 nM。 |
|
|
| S1460 |
SP600125
|
SP600125 (Nsc75890)是一种广谱JNK抑制剂,作用于JNK1、JNK2和JNK3,无细胞试验中IC50分别为40 nM、40 nM和90 nM,比作用于MKK4选择性高10倍,比作用于MKK3、MKK6、PKB和PKCα选择性高25倍,比作用于ERK2、p38、Chk1、EGFR等选择性高100倍。SP600125也是一种广谱的serine/threonine kinases抑制剂,包括Aurora kinase A、FLT3和TRKA,对应的IC50值为60 nM、90 nM和70 nM。SP600125可抑制自噬而激活细胞凋亡。 |
- Research (Wash D C), 2026, 9:1190
- Anal Chem, 2026, 98(10):7247-7261
- iScience, 2026, 29(5):115396
|

|
| S7397 |
Sorafenib (BAY 43-9006)
|
Sorafenib是Raf-1和B-Raf的多重激酶抑制剂,无细胞试验中IC50分别为6 nM和22 nM。Sorafenib 还可抑制VEGFR-2、VEGFR-3、PDGFR-β、Flt-3和c-KIT,对应的IC50值分别为90 nM、20 nM、57 nM、59 nM和68 nM。Sorafenib 可诱导autophagy、apoptosis并激活ferroptosis,Sorafenib 具有抗肿瘤活性。 |
- Signal Transduct Target Ther, 2026, 11(1)79
- Int J Surg, 2026, 10.1097/JS9.0000000000004913
- Acta Pharmacol Sin, 2026, 10.1038/s41401-026-01791-z
|

|
| S1040 |
Sorafenib Tosylate (BAY 43-9006)
|
Sorafenib tosylate是Raf-1和B-Raf的多重激酶抑制剂,无细胞试验中IC50分别为6 nM和22 nM。Sorafenib 还可抑制VEGFR-2、VEGFR-3、PDGFR-β、Flt-3和c-KIT,对应的IC50值分别为90 nM、20 nM、57 nM、59 nM和68 nM。Sorafenib Tosylate 可诱导autophagy、apoptosis并激活ferroptosis,并具有抗肿瘤活性。 |
- Int J Oncol, 2025, 67(3)72
- Nature, 2024, 629(8013):927-936
- Cell Mol Life Sci, 2024, 81(1):238
|

|
| S1526 |
Quizartinib (AC220)
|
Quizartinib (AC220)是一种二代FLT3抑制剂,作用于Flt3(ITD/WT),在MV4-11和 RS4;11细胞中IC50分别为1.1 nM/4.2 nM,作用于Flt3比作用于KIT,PDGFRA,PDGFRB,RET,和CSF-1R选择性高10倍。Quizartinib (AC220)可诱导肿瘤细胞的凋亡。Phase 3。 |
- Cell Rep Med, 2026, 7(3):102687
- Cell Death Discov, 2026, 12(1):98
- Signal Transduct Target Ther, 2025, 10(1):222
|

|
| S1048 |
Tozasertib (VX-680, MK-0457)
|
Tozasertib (VX-680)是一种pan-Aurora抑制剂,对Aurora A作用最强,无细胞试验中Kiapp为0.6 nM,而对Aurora B/Aurora C的作用较弱,对Aurora A选择性比其他55种激酶高100倍。唯二的例外是 Fms-related tyrosine kinase-3 (FLT-3) 和 BCR-ABL tyrosine kinase,也可被 Tozasertib 抑制,对应的Ki值均为30 nM和30 nM。Tozasertib 可诱导凋亡和自噬。Phase 2。 |
- bioRxiv, 2026, 2026.01.11.698900
- ACS Appl Bio Mater, 2026, 9(8):3753-3767
- Cell Rep Med, 2025, S2666-3791(25)00102-8
|

|
| S7818 |
Pexidartinib (PLX3397)
|
Pexidartinib (PLX3397)是一种口服有效的多靶点CSF-1R,Kit (c-Kit),和 FLT3受体酪氨酸激酶抑制剂,其IC50分别为20 nM, 10 nM 和 160 nM。Pexidartinib (PLX3397) 可以诱导细胞凋亡和坏死,并具有抗肿瘤活性。Phase 3。 |
- Nature, 2026, 652(8109):442-450
- Brain Behav Immun, 2026, 136:106576
- Oncogene, 2026, 45(21):1970-1987.
|

|
| S2194 |
R406
|
R406(R406 besylate) 是一种有效的 Syk 抑制剂,无细胞试验中IC50为41 nM,对Syk抑制作用强,但是不抑制Lyn,对Flt3的作用比对Syk低5倍。R406 可诱导凋亡。Phase 1。 |
- Aging (Albany NY), May 7, 2020, 8221-8240
- eLife, April 30, 2020, e56656
- Cancer Research, April 15, 2017, 1818-1830
|

|
| S7083 |
Ceritinib (LDK378)
|
Ceritinib是一种有效的ALK抑制剂,在无细胞试验中IC50为0.2 nM。Ceritinib (LDK378)还可抑制IGF-1R、InsR、STK22D和FLT3对应的IC50值分别为8 nM、7 nM、23 nM和60 nM。Phase 3。 |
- Molecular Cancer Therapeutics, January 01 2018, 175-186
- Oncotarget, December 29 2015, 44643-44659
- ESMO Open, August 2021, 100172
|

|
| S1533 |
R406 (free base)
|
R406 (free base) 是一种有效的Syk抑制剂,无细胞试验中IC50为41 nM,强效抑制Syk而不能抑制Lyn,对Flt3作用效果低5倍。R406 (free base) 可触发凋亡。Phase 1。 |
- Journal of Leukocyte Biology, July 25, 2024, 210-223
- PLoS One, January 22, 2019, e0210879
- Blood, February 11, 2016, 739-48
|

|
| S4001 |
Cabozantinib S-malate
|
Cabozantinib malate (XL184)是Cabozantinib的苹果酸盐,是有效的VEGFR2抑制剂,IC50为0.035 nM,也抑制c-Met, RET (c-RET), Kit (c-Kit), Flt-1/3/4, Tie2和AXL,无细胞试验中IC50分别为1.3 nM, 4 nM, 4.6 nM, 12 nM/11.3 nM/6 nM, 14.3 nM和7 nM。Cabozantinib malate (XL184)可诱导细胞凋亡。 |
- Scientific Reports, 2025, 35889
- Sci Rep, 2025, 15(1):35889
- bioRxiv, 2025, 2025.08.15.670608
|

|
| S2736 |
Fedratinib (TG101348)
|
Fedratinib (SAR302503, TG101348)是一种选择性JAK2抑制剂,在无细胞试验中IC50为3 nM,作用于JAK2比作用于JAK1和JAK3选择性高35和334倍。Fedratinib也可抑制 FMS-like tyrosine kinase 3 (FLT3) 和 Ret (c-RET),对应的IC50值分别为15 nM和48 nM。Fedratinib有潜在的抗肿瘤活性。Fedratinib可抑制细胞增殖并促进凋亡。Phase 2。 |
- Nat Commun, 2026, 17(1):949
- Leukemia, 2025, 39(7):1678-1691
- Front Immunol, 2025, 16:1514618
|

|
| S2730 |
Crenolanib (CP-868596)
|
Crenolanib是一种有效的,选择性PDGFRα/β抑制剂,在CHO细胞中Kd为2.1 nM/3.2 nM,也能有效抑制FLT3,对D842V突变型敏感对V561D突变型不敏感,作用于PDGFR比作用于c-Kit,VEGFR-2,TIE-2,FGFR-2,EGFR,erbB2,和Src的选择性高100倍以上。Crenolanib可辅助诱导线粒体自噬。 |
- Exp Mol Pathol, 2026, 146:105037
- Cancer Cell, 2025, S1535-6108(25)00070-4
- Nat Commun, 2025, 16(1):1358
|

|
| S1111 |
Foretinib (GSK1363089, XL880)
|
Foretinib是一种ATP竞争性的HGFR和VEGFR抑制剂,对Met (c-Met)和KDR作用最强,在无细胞试验中IC50分别为0.4 nM和0.9 nM。对Ron, Flt-1/3/4, Kit (c-Kit), PDGFRα/β和Tie-2作用效果稍弱,对FGFR1和EGFR几乎没有抑制活性。Phase 2。 |
- J Microbiol, 2025, 63(2):e2409001
- Int J Mol Sci, 2023, 24(1)757
- Biol Open, 2023, 12(8)bio059994
|

|
| S7754 |
ASP2215 (Gilteritinib)
|
Gilteritinib (ASP2215)是小分子FLT3/AXL抑制剂,对FLT3和AXL的IC50值为0.29 nM和0.73 nM。其抑制c-KIT的IC50值约为抑制FLT3的800倍。 |
- Heliyon, October 12, 2022, e11004
- bioRxiv, November 24, 2025, nan
- Cell Death Discov, 2026, 12(1):98
|
|
| S1018 |
Dovitinib (TKI-258)
|
Dovitinib (TKI-258, CHIR-258)是一种多靶点的RTK抑制剂,在无细胞试验中对III型(FLT3/c-Kit)作用最强,IC50为1 nM/2 nM,同时也作用于IV类(FGFR1/3)和V类(VEGFR1-4) RTKs,IC50为8-13 nM,但对InsR,EGFR,c-Met,EphA2,Tie2,IGF-1R和HER2作用较弱。Phase 4。 |
- Acta Pharmacol Sin, 2026, 10.1038/s41401-026-01791-z
- Spectrochim Acta A Mol Biomol Spectrosc, 2025, 343:126602
- Biomed Pharmacother, 2024, 180:117533
|

|
| S8032 |
PRT062607 (P505-15) HCl
|
PRT062607 (P505-15, BIIB057, PRT-2607)是一种新型的,高度选择性Syk抑制剂,无细胞试验中IC50为1 nM,作用于Syk比作用于Fgr,PAK5,Lyn,FAK,Pyk2, FLT3, MLK1 和Zap70选择性高80倍以上。 |
- Sci Adv, 2025, 11(31):eadu4270
- Scientific Reports, 2024, 7739
- Sci Rep, 2024, 14(1):7739
|

|
| S8229 |
Brigatinib
|
Brigatinib是一种有效的、选择性的ALK抑制剂,IC50=0.6 nM;也是ROS1抑制剂,IC50=0.9 nM。它还能以相对较低的效力抑制IGF-1R、FLT3、FLT3(D835Y)和EGFR。 |
- Leukemia, 2025, 10.1038/s41375-025-02682-8
- Cell Death Dis, 2025, 16(1):194
- Toxicol Appl Pharmacol, 2025, 498:117310
|

|
| S2198 |
SGI-1776 free base
|
SGI-1776 free base是一种新型的,ATP竞争性Pim1抑制剂,无细胞试验中IC50为7 nM,比作用于Pim2和Pim3选择性分别高50和10倍,也有效作用于Flt3和haspin。SGI-1776 可诱导凋亡和自噬。 |
- Cell Death Dis, 2024, 15(9):644
- Mar Drugs, 2024, 22(10)444
- Mol Cancer, 2023, 22(1):64
|

|
| S7119 |
Go 6976
|
Go6976 (PD406976)是一种强效的PKC抑制剂,其对PKC (老鼠大脑), PKCα, and PKCβ1的IC50分别为7.9nM,2.3 nM, 和 6.2 nM 。此外,也是JAK2 和 Flt3的强抑制剂。
|
- FASEB J, 2025, 39(12):e70768
- bioRxiv, 2025, 2025.02.02.636139
- Int J Biol Sci, 2024, 20(10):3956-3971
|

|
| S1003 |
Linifanib (ABT-869)
|
Linifanib (ABT-869, AL39324, RG3635)是一种新型有效的ATP竞争性VEGFR/PDGFR抑制剂,作用于KDR,CSF-1R,Flt-1/3和PDGFRβ,其IC50分别为4 nM,3 nM,3 nM/4 nM和66 nM,对突变激酶依赖性癌细胞(即FLT3)最有效。Linifanib (ABT-869) 可诱导自噬和凋亡。Phase 3。 |
- Acta Pharmacol Sin, 2026, 10.1038/s41401-026-01791-z
- Research in Pharmaceutical Sciences, 2025 Oct 20, 734-745
- Biomed Pharmacother, 2024, 180:117533
|

|
| S7765 |
Dovitinib (TKI258) Lactate monohydrate
|
Dovitinib (TKI258) Lactate monohydrate是Dovitinib的乳酸盐,Dovitinib是一种多靶点的PTK抑制剂,主要对第III类(FLT3/c-Kit)发挥作用,IC50为1 nM/2 nM,对第IV 类(FGFR1/3)和第V 类(VEGFR1-4) RTKs也有效,IC50为8-13 nM,对InsR,EGFR,c-Met,EphA2,Tie2,IGFR1 和HER2作用较差。Phase 4。
|
- Mol Cell, 2021, S1097-2765(21)00507-4
- Acta Neuropathol, 2021, 10.1007/s00401-021-02327-x
- Cancer Res, 2021, canres.1780.2020
|

|
| S8057 |
Pacritinib (SB1518)
|
Pacritinib是有效的选择性Janus Kinase 2 (JAK2)和Fms-Like Tyrosine Kinase-3 (FLT3)抑制剂,无细胞试验中IC50分别为23和22 nM。Phase 3。 |
- Nat Commun, 2026, 17(1):949
- Front Pharmacol, 2025, 16:1549183
- Toxicol In Vitro, 2025, S0887-2333(25)00050-5
|

|
| S1244 |
Amuvatinib (MP-470)
|
Amuvatinib (MP-470, HPK 56) 是一种有效的,作用于c-Kit、PDGFα和Flt3的多靶点抑制剂,IC50分别为10 nM、40 nM和81 nM。Amuvatinib 可抑制c-MET和c-RET。Amuvatinib 还具有DNA修复蛋白Rad51抑制剂及抗肿瘤的活性。Phase 2。 |
- Cancer Res, 2025, 10.1158/0008-5472.CAN-24-2220
- Hematol Oncol, 2025, 43(5):e70131
- bioRxiv, 2024, 2023.11.21.568071
|

|
| S2692 |
TG101209
|
TG101209是一种选择性的JAK2抑制剂,无细胞试验中IC50为6 nM,对Flt3和RET (c-RET)作用效果稍弱,IC50分别为25 nM和17 nM,作用于JAK2比作用于JAK3选择性高30倍左右,对JAK2V617F和MPLW515L/K突变型敏感。 |
- Signal Transduct Target Ther, 2025, 10(1):131
- Phytomedicine, 2023, 117:154918
- EBioMedicine, 2022, 78:103963
|

|
| S1043 |
Tandutinib (MLN518)
|
Tandutinib (MLN518, CT53518, NSC726292)是一种有效的FLT3拮抗剂,IC50为0.22 μM,同时也抑制PDGFR和c-Kit,对FLT3作用效果比CSF-1R强15到20倍,比FGFR, EGFR和KDR等强100倍以上。Phase 2。 |
- Cancer Cell, 2021, S1535-6108(21)00659-0
- Drug Metab Dispos, 2021, 49(1):53-61
- Genome Med, 2020, 18;12(1):17
|

|
| S1181 |
ENMD-2076
|
ENMD-2076 选择性地作用于 Aurora A 和 Flt3,IC50分别为14 nM和1.86 nM,作用于Aurora A比作用于Aurora B选择性高25倍,对RET、SRC、NTRK1/TRKA、CSF1R/FMS、VEGFR2/KDR、FGFR和PDGFRα作用效果稍弱。ENMD-2076 可抑制各种人类实体瘤和造血癌细胞系的生长,其IC50值为0.025至0.7μM,可诱导凋亡和G2/M期停滞。Phase 2。 |
- Int J Biol Macromol, 2025, 292:139119
- Cell, 2021, 184(2):334-351.e20
- Cancer Discovery, 2021, 2544-2563
|

|
| S2158 |
KW-2449
|
KW-2449是一种多靶点抑制剂,最有效作用于Flt3,IC50为6.6 nM,作用于Flt3, Bcr-Abl和Aurora A适度有效;对PDGFRβ, IGF-1R, EGFR没有抑制效果。Phase 1。 |
- Molecular Cancer Research, October 2018, 1556-1567
- Cancer Research, June 6, 2022, 2141-2155
- Stem Cell Research & Therapy, December 27, 2022, 534
|

|
| S7576 |
UNC2025 HCl
|
UNC2025 HCl 是一种有效且口服生物可利用的双重MER/FLT3抑制剂,IC50分别为0.74 nM 和 0.8 nM,选择性比作用于Axl和 Tyro3大约高20倍。
|
- bioRxiv, 2025, 2025.07.18.665641
- Immunity, 2023, 56(8):1778-1793.e10
- International Journal of Molecular Sciences, 2023, 15903
|
|
| S7014 |
Merestinib (LY2801653)
|
Merestinib (LY2801653) 是一种2型ATP竞争型的慢抑制剂,抑制Met (c-Met)酪氨酸激酶,Ki值为2 nM。药效的停留时间为0.00132 min(-1),t1/2为525 min。Merestinib (LY2801653) 还可抑制MST1R、AXL、ROS1、MKNK1/2、FLT3、MERTK、DDR1和DDR2,其对应的IC50值分别为11 nM、2 nM、23 nM、7 nM、7 nM、10 nM、0.1 nM 和 7 nM。 |
- Mol Oncol, 2025, 19(8):2366-2387
- Molecular Oncology, 2025, 2366-2387
- NPJ Breast Cancer, 2024, 10(1):65
|
|
| S8442 |
TAK-659 Hydrochloride
|
TAK-659 Hydrochloride是一种有效的、选择性的SYK抑制剂。IC50为3.2 nM。对其他激酶具有选择性,对FLT3也能有效抑制,IC50为4.6 nM。 |
- ACS Pharmacology & Translational Science, May 14, 2025, 615160
- Experimental and Therapeutic Medicine, April 4, 2025, 110
- Metabolomics, 2026, 22(3)51
|
|
| S9662 |
UNC2025
|
UNC2025 是一种有效的、口服活性的 FLT3 和 MER 的双重抑制剂,对应的IC50值分别为0.35 nM和0.46 nM。UNC2025 还抑制 AXL、TRKA、TRKC、QIK、TYRO3、SLK、NuaK1、Kit (c-Kit) 和 Met (c-Met),对应的IC50值分别为1.65 nM、1.67 nM、4.38 nM、5.75 nM、5.83 nM、6.14 nM、7.97 nM、8.18 nM 和 364 nM。 |
- iScience, 2024, 27(7):110226
- Commun Biol, 2023, 6(1):916
- Int J Mol Sci, 2023, 10.3390/ijms242115903
|
|
| S7003 |
AZD2932
|
AZD2932 是一种有效的多靶点蛋白酪氨酸激酶抑制剂,对VEGFR-2,PDGFRβ,Flt-3,和 c-Kit 的 IC50 分别为 8 nM,4 nM,7 nM,和 9 nM。
|
- Cancer Research, 2021, 747-762
- Cancer Res, 2020, canres.1992.2020
- Molecules, 2020, 8;25(9) pii: E2220
|
|
| S0278 |
SU5614
|
SU5614 (Chloro-SU5416, Chloro-Semaxanib) 是一种小分子 receptor tyrosine kinases (RTK) 的抑制剂,对 VEGFR-2、c-kit 以及野生型和突变型 FLT3 均有抑制作用。SU5614 可减少细胞增殖并诱导凋亡。 |
- Frontiers in Cell and Developmental Biology, January 26, 2022, 797047
- Advanced Science, August 7, 2021, e2101848
- Am J Pathol, 2024, S0002-9440(24)00326-2
|
|
| S6662 |
AST-487 (NVP-AST487)
|
AST-487 (NVP-AST487),一种N,N'-二苯基脲,是Flt3的竞争性抑制剂,ki值为0.12 μM。除FLT3以外,AST487还抑制RET,KDR,c-KIT 和 c-ABL 激酶,IC50值低于1 μM。 |
- Cancer Cell, 2022, S1535-6108(22)00312-9
- International Journal of Molecular Sciences, 2022, 10819
- Int J Mol Sci, 2022, 23(18)10819
|
|
| S8023 |
TCS 359
|
TCS 359是一种有效的FLT3抑制剂,IC50为42 nM。 |
- Nat Commun, 2021, 12(1):2148
- Blood, 2016, 127(23):2890-90
|
|
| S2018 |
ENMD-2076 L-(+)-Tartaric acid
|
ENMD-2076 L-(+)-Tartaric acid 是ENMD-2076的酒石酸,选择性作用于Aurora A和VEGFR(Flt3),IC50分别为14 nM和1.86 nM,作用于Aurora A比作用于Aurora B选择性高25倍,对VEGFR2/KDR和VEGFR3, FGFR1和FGFR2和PDGFRα作用效果稍弱。Phase 2。 |
- Veterinary and Comparative Oncology, 2016 Mar, 14(1):13-27
- International Journal of Oncology, 2015 Jul, 47(1):122-32
|
|
| S8899 |
FF-10101
|
FF-10101是一种新型的不可逆的FLT3抑制剂,对FLT3(WT)和 FLT3(D835Y)的ic50分别为0.20 nM 和 0.16 nM。 |
- Frontiers in Oncology, August 20, 2021, 686765
- ACS Pharmacology & Translational Science, 2025, nan
- Frontiers in Oncology, 2021, 686765
|
|
| S6839 |
MRX-2843
|
MRX-2843 (UNC2371) 是酪氨酸激酶 MERTK 和 FLT3 的口服活性双重抑制剂,对应的IC50值分别为1.3 nM和0.64 nM。 |
- Cancer Immunology Research, 2025, CIR-24-0335
- Scientific Reports, 2024, 4125
- Scientific Reports, 2024, 14:4125
|
|
| S7545 |
G-749
|
G-749是一种新型有效的FLT3抑制剂,对FLT3(WT),FLT3(D835Y),和Mer的IC50分别为0.4 nM,0.6nM 和 1nM,对其它酪氨酸激酶效果较低。
|
- Scientific Reports, 2024, 4125
- Front Pharmacol, 2021, 12:730241
|
|
| E2387 |
FLT3-IN-2
|
FLT3-IN-2是一种FLT3抑制剂,IC50低于1 μM。 |
- Pathogens, 2022, 12(1)53
|
|
| S9779 |
Emavusertib (CA-4948)
|
Emavusertib (CA-4948) 是一种有效的、口服生物可利用的 interleukin-1 receptor-associated kinase 4/FMS-like Tyrosine Kinase 3 (IRAK4/FLT3) 的抑制剂,具有抗肿瘤活性。 |
- Arthritis Rheumatol, 2021, 10.1002/art.42043
|
|
| S1099 |
SKLB4771 (FLT3-IN-1)
|
SKLB4771是一种有效、选择性的人类受体型酪氨酸蛋白激酶FLT3抑制剂,IC50为10 nM。 |
- Front Pharmacol, 2022, 13:899775
|
|
| S7533 |
AMG 925
|
AMG 925是一种有效的,且口服具有生物活性的双重FLT3/CDK4抑制剂,IC50分别为1 nM 和 3 nM。
|
- Molecules, 2020, 8;25(9) pii: E2220
|
|
| S7002 |
Zotiraciclib
|
Zotiraciclib (SB1317; TG02)是一种新型小分子CDK/JAK2/FLT3抑制剂,对CDK1、2和9的IC50分别为9、5和3 nM,抑制FLT3的IC50为19 nM,抑制JAK2的IC50为19 nM。 |
|
|
| E1437 |
PHI-101
|
PHI-101 是一种口服的强效第三代 FLT3 抑制剂,可克服对多种耐药突变的耐药性。它具有研究复发或难治性急性髓系白血病 (AML) 的潜力。 |
|
|
| S0550 |
5'-Fluoroindirubinoxime
|
5'-Fluoroindirubinoxime (5’-FIO, compound 13)是一种靛红素的衍生物,也是一种有效的FMS-样酪氨酸激酶3(FMS-like tyrosine kinase 3,FLT3)抑制剂,IC50为15 nM。 |
|
|
| E4598 |
MEN1703(SEL24,SEL24-B489)
|
MEN1703(SEL24-B489,SEL24, Pim/flt3 kinase inhibitor SEL24) 是一种有效的 I 型口服 PIM 和 FLT3-ITD 双重抑制剂,其对 PIM1 的 Kd 值为 2 nM,对 PIM2 的 Kd 值为 2 nM,对 PIM3 的 Kd 值为 3 nM。MEN1703 在治疗急性髓系白血病方面表现出广泛的治疗潜力。 |
|
|
| S9275 |
Isoguanosine
|
Isoguanosine (Crotonoside)抑制 FLT3 和 HDAC3/6 治疗AML。Isoguanosine 是巴豆(Croton tiglium)种子中天然存在的鸟苷的活性异构体。 |
|
|
| S7640 |
Lestaurtinib
|
Lestaurtinib (CEP-701, KT-5555) 是 indolocarbazole K252 的衍生物,也是星形孢菌素类似物。它是一种口服的受体蛋白酪氨酸激酶 (RPTK) 抑制剂,可抑制 FLT3 和 Trk,IC50 分别为 3 nM 和 < 25 nM。它还能抑制 JAK2,IC50 为 0.9 nM。 |
|
|
| S5986 |
FLT3-IN-3
|
FLT3-IN-3 是 FLT3 的强效抑制剂,对 FLT3 WT 和 FLT3 D835Y 的 IC50 值分别为 13 nM 和 8 nM。FLT3-IN-3 在低纳摩尔浓度下可有效抑制 FLT3-ITD 阳性 MV4-11 和 MOLM-13 细胞系的增殖。 |
|
|
| E4720 |
Flonoltinib
|
Flonoltinib (JAK2/FLT3-IN-1) 是一种强效且选择性的JAK2和FLT3双重抑制剂,体外激酶试验中,对JAK2、JAK2V617F和FLT3的IC50分别为0.8、1.4和15 nM。它还能抑制细胞增殖,并诱导噬血细胞淋巴组织细胞增生症(HLH)患者CD8+T细胞、CTLL2细胞和RAW 264.7细胞的G2/M细胞周期停滞和凋亡。 |
|
|
| S6060 |
HPK1-IN-2
|
HPK1-IN-2 是一种有效的和口服活性的 hematopoietic progenitor kinase-1 (HPK1, a serine/threonine Ste20-related protein kinase)、Lck 和 Flt3 的抑制剂,其IC50值分别为 <0.05 μM、<0.5 μM 和 <0.05 μM。HPK1-IN-2 具有抗肿瘤的活性。 |
|
|
| E2658 |
FN-1501
|
FN-1501是Fms样受体酪氨酸激酶3 (Fms-like receptor tyrosine kinase 3, FLT3) 和细胞周期蛋白依赖性激酶(cyclin-dependent kinase, CDK)的有效抑制剂,对CDK2/cyclin A、CDK4/cyclin D1、CDK6/cyclin D1和FLT3的IC50分别为2.47、0.85、1.96和0.28 nM。 |
|
|
| S9892 |
HM43239
|
HM43239 是一种新型强效小分子 FLT3 抑制剂,不仅选择性抑制 FLT3 野生型、ITD 突变体或 TKD 突变,还选择性抑制 FLT3 ITD/TKD 双突变。 HM43239 对 FLT3 WT、FLT3 ITD 和 FLT3 D835Y 激酶的 IC50 分别为 1.1 nM、1.8 nM 和 1.0 nM。 |
|
|


























