| S1025 |
Gefitinib (ZD1839)
|
Gefitinib是一种EGFR抑制剂,作用于NR6wtEGFR和NR6W细胞中的Tyr1173、Tyr992、Tyr1173和Tyr992,IC50 分别为37 nM、37nM、26 nM和57 nM。Gefitinib 可通过阻滞PI3K/AKT/mTOR信号通路来促进肺癌细胞的自噬和细胞凋亡。 |
-
Nat Commun, 2025, 16(1):7287
-
Nat Commun, 2025, 16(1):6451
-
Cell Rep Med, 2025, 6(2):101929
|

|
| S7786 |
Erlotinib (CP-358774)
|
Erlotinib是一种EGFR抑制剂,IC50 为 2 nM,对EGFR的敏感性比对人c-Src 或 v-Ab高1000多倍。Erlotinib可诱导自噬。 |
-
Nat Commun, 2025, 16(1):8932
-
Cell Rep Med, 2025, S2666-3791(25)00272-1
-
J Exp Clin Cancer Res, 2025, 44(1):290
|
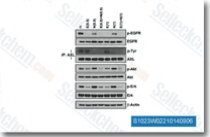
|
| S1023 |
Erlotinib (CP-358774) Hydrochloride
|
Erlotinib HCl 是一种EGFR抑制剂,在无细胞试验中IC50为2 nM,对EGFR的选择性比对人c-Src或v-Abl高1000多倍。 |
-
Nat Commun, 2026, 17(1)3228
-
Cell Res, 2025, 10.1038/s41422-025-01110-x
-
Nat Commun, 2025, 16(1):3591
|
|
| S2111 |
Lapatinib (GW572016)
|
Lapatinib 是一种有效的EGFR和ErbB2抑制剂,在无细胞试验中IC50分别为10.2和9.8 nM。Lapatinib 可诱导 ferroptosis 和细胞自噬。 |
-
Cells, 2026, 15(4)383
-
Nat Genet, 2025, 57(6):1452-1462
-
Cancer Discov, 2025, 10.1158/2159-8290.CD-25-0605
|

|
| S7297 |
AZD9291 (Osimertinib)
|
Osimertinib (AZD9291, Mereletinib)是口服不可逆的,突变选择性EGFR抑制剂,在 LoVo细胞中对Exon 19 缺失的 EGFR,L858R/T790M EGFR,和 WT EGFR的IC50分别为12.92,11.44 和 493.8 nM。Phase 3。 |
-
Acta Pharmacologica Sinica, November 15, 2017, 1512-1520
-
Clinical Cancer Research, July 01, 2018, 3097-3107
-
Cancer Biology & Medicine, December 09, 2024, 1156-1170
|

|
| S1011 |
Afatinib (BIBW2992)
|
Afatinib不可逆地抑制EGFR/ErbB,包括EGFR(wt),EGFR(L858R),EGFR(L858R/T790M),ErbB2(HER2)和ErbB4(HER4),在无细胞试验中IC50分别为0.5 nM,0.4 nM,10 nM,14 nM和1 nM。Afatinib可诱导自噬。 |
-
Cell Res, 2025, 10.1038/s41422-025-01110-x
-
Nat Genet, 2025, 10.1038/s41588-025-02158-6
-
Cell Rep Med, 2025, 6(2):101929
|

|
| S1006 |
Saracatinib (AZD0530)
|
Saracatinib (AZD0530)是一种有效的Src抑制剂,无细胞试验中IC50为2.7 nM,对c-Yes, Fyn, Lyn, Blk, Fgr和Lck也具有活性;但对Abl和EGFR (L858R和L861Q)活性较低。Saracatinib可诱导自噬。Phase 2/3。 |
-
Cancers (Basel), 2026, 18(7)1082
-
J Clin Invest, 2025, e177599
-
Cell Rep, 2025, 44(1):115109
|
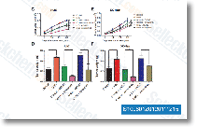
|
| S5098 |
Gefitinib hydrochloride
|
Gefitinib (ZD1839) is an EGFR inhibitor with IC50s of 15.5 nM and 823.3 nM for WT EGFR and EGFR (858R/T790M), respectively. |
-
Cell Death Dis, 2024, 15(8):555
-
iScience, 2024, 27(10):110862
-
Cancer Res, 2023, 83(2):316-331
|
|
| S7810 |
Afatinib Dimaleate
|
Afatinib Dimaleate不可逆地抑制 EGFR/HER2,包括 EGFR(wt),EGFR(L858R),EGFR(L858R/T790M) 和 HER2,IC50 分别为0.5 nM,0.4 nM,10 nM 和 14 nM;活性比对Gefitinib抵抗型L858R-T790M EGFR突变体高100倍。Afatinib (BIBW2992) Dimaleate 可诱导自噬。 |
-
Cell, 2024, 187(3):712-732.e38
-
Signal Transduct Target Ther, 2024, 9(1):153
-
J Invest Dermatol, 2024, S0022-202X(23)03210-4
|
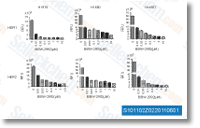
|
| S1028 |
Lapatinib Ditosylate
|
Lapatinib Ditosylate是一种有效的EGFR 和 ErbB2抑制剂,在无细胞试验中IC50分别为10.8和9.2 nM。 |
-
Cell Death Dis, 2025, 16(1):118
-
bioRxiv, 2025, 2025.05.29.656249
-
NPJ Breast Cancer, 2024, 10(1):65
|
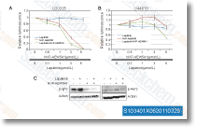
|
| S1143 |
AG-490
|
AG-490是一种EGFR抑制剂,在无细胞试验中IC50为0.1 μM,作用于EGFR比作用于ErbB2选择性高135倍,对JAK2也有抑制作用,对Lck,Lyn,Btk,Syk和Src没有抑制活性。 |
-
Biomed Pharmacother, 2024, 180:117568
-
Commun Biol, 2024, 7(1):1694
-
NPJ Breast Cancer, 2024, 10(1):80
|

|
| S1046 |
Vandetanib (ZD6474)
|
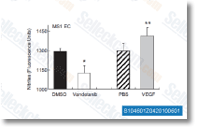
Vandetanib是一种有效的 VEGFR2 抑制剂,在无细胞试验中IC50为40 nM。同时,也抑制VEGFR3和EGFR,IC50分别为110 nM 和500 nM。对PDGFRβ, Flt-1, Tie-2 和FGFR1作用效果不大,IC50为 1.1-3.6 μM, 对MEK, CDK2, c-Kit, erbB2, FAK, PDK1, Akt 和 IGF-1R几乎没有作用效果,IC50>10 μM。Vandetanib (ZD6474) 可增加凋亡并通过提高 reactive oxygen species (ROS) 的水平来诱导自噬。 |
-
Frontiers in Pharmacology, May 10, 2024, 15:1345070
-
Molecular Cancer Therapeutics, March 15, 2016, 503-11
-
Oncotarget, June 14, 2016, 36101-36114
|
|
| S2150 |
Neratinib (HKI-272)
|
Neratinib是一种高度选择性的HER2和EGFR抑制剂,在无细胞试验中IC50分别为59 nM 和 92 nM;微弱抑制KDR和Src,对Akt,CDK1/2/4,IKK-2,MK-2,PDK1,c-Raf和c-Met没有显著的抑制作用。Phase 3。 |
-
Nat Genet, 2025, 57(6):1452-1462
-
Cancer Discov, 2025, 10.1158/2159-8290.CD-25-0605
-
Neoplasia, 2025, 70:101246
|
|
| S1167 |
CP-724714
|
CP-724714是一种有效的,选择性HER2/ErbB2抑制剂,IC50为10 nM,无细胞试验中比作用于EGFR,InsR,IRG-1R,PDGFR,VEGFR2,Abl,Src,c-Met等选择性高640多倍。Phase 2。 |
-
Front Immunol, 2024, 15:1335302
-
Cells, 2024, 13(17)1452
-
Cancer Res Commun, 2024, 10.1158/2767-9764.CRC-24-0221
|

|
| S5078 |
Osimertinib mesylate
|
Osimertinib mesylate (AZD9291) 是osimertinib的甲磺酸盐形式,是口服的第三代 epidermal growth factor receptor (EGFR) 酪氨酸激酶(TKI)抑制剂。 |
-
Drug Resist Updat, 2025, 81:101237
-
Oncogene, 2024, 10.1038/s41388-024-03220-z
-
iScience, 2024, 27(10):110862
|
|
| S2728 |
AG-1478
|
AG-1478是一种选择性EGFR抑制剂,在无细胞试验中IC50为3 nM,对HER2-Neu,PDGFR,Trk,Bcr-Abl和InsR几乎没有作用活性。AG-1478(Tyrphostin AG-1478)通过靶向 phosphatidylinositol 4-kinase IIIα (PI4KA) 抑制脑心肌炎病毒encephalomyocarditis virus (EMCV)和丙型肝炎病毒hepatitis c virus (HCV)。 |
-
PLoS Negl Trop Dis, 2026, 20(3):e0014153
-
Cell Death Dis, 2025, 16(1):479
-
J Stroke Cerebrovasc Dis, 2025, 34(6):108315
|

|
| S2727 |
Dacomitinib (PF-299804)
|
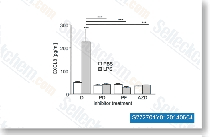
Dacomitinib是一种有效的,不可逆的泛ErbB抑制剂,最有效作用于EGFR,无细胞试验中IC50为6 nM。Dacomitinib 抑制 ERBB2 和 ERBB4 ,其对应的IC50值分别为45.7 nM和73.7 nM。Dacomitinib 可高效作用于携带EGFR或ERBB2突变型(耐Gefitinib)和携带EGFR T790M突变型的NSCLCs。Dacomitinib 可抑制细胞生长并诱导凋亡。Phase 2。 |
-
Cell Rep Med, 2025, 6(8):102284
-
Cell Oncol (Dordr), 2025, 10.1007/s13402-025-01097-y
-
Nat Commun, 2024, 15(1):2742
|
|
| S1019 |
Canertinib (CI-1033)
|
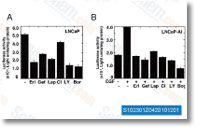
Canertinib (CI-1033, PD183805)是一种泛ErbB抑制剂,作用于EGFR和ErbB2,IC50分别为1.5 nM和9.0 nM,但对PDGFR,FGFR,InsR,PKC,和CDK1/2/4等均无抑制活性。Phase 3。 |
-
Journal of Cell Science, September 15, 2010, 3102-3111
-
Cancer Research, March 1, 2021, 1398-1412
-
Cells, January 26, 2022, 425
|
|
| S2192 |
Sapitinib (AZD8931)
|
Sapitinib (AZD8931)是一种可逆的,ATP竞争性EGFR,ErbB2和ErbB3抑制剂,无细胞试验中IC50分别为4 nM,3 nM和4 nM,作用于NSCLC细胞更有效,作用于ErbB家族比作用于MNK1和Flt选择性强100倍。Phase 2。 |
-
Cell Death Dis, 2025, 16(1):457
-
Biomolecules, 2025, 15(5)698
-
Cell Death Dis, 2024, 15(12):912
|
|
| S8229 |
Brigatinib
|
Brigatinib是一种有效的、选择性的ALK抑制剂,IC50=0.6 nM;也是ROS1抑制剂,IC50=0.9 nM。它还能以相对较低的效力抑制IGF-1R、FLT3、FLT3(D835Y)和EGFR。 |
-
Leukemia, 2025, 10.1038/s41375-025-02682-8
-
Cell Death Dis, 2025, 16(1):194
-
Toxicol Appl Pharmacol, 2025, 498:117310
|

|
| S2250 |
EGCG ((-)-Epigallocatechin Gallate)
|
EGCG ((-)-Epigallocatechin Gallate)是从绿茶中分离的一种有效的抗氧化剂Polyphenol flavonoid。抑制telomerase和DNA methyltransferase, 阻滞EGF受体和HER-2受体的激活,抑制脂肪酸合成酶以及谷氨酸脱氢酶的活性。 |
-
Clin Mol Hepatol, 2025, 10.3350/cmh.2024.0694
-
Cell Rep, 2025, 44(6):115799
-
Front Pharmacol, 2024, 15:1403424
|

|
| S1342 |
Genistein
|
Genistein是一种来源于大豆的植物雌激素,是一种高特异性的蛋白酪氨酸激酶(PTK) 的抑制剂,在NIH-3T3细胞中通过EGF或者胰岛素介导而抑制有丝分裂,IC50分别为12μM和19 μM。 |
-
J Immunother Cancer, 2025, 13(6)e011442
-
Cell Rep, 2025, 44(6):115849
-
International Journal of Polymeric Materials and Polymeric Biomaterials, 2025, 1563-1572
|
|
| S7284 |
Rociletinib (CO-1686)
|
Rociletinib (CO-1686, AVL-301)是一种不可逆的,选择性抑制突变型EGFR,在无细胞试验中作用于EGFRL858R/T790M和EGFRWT, Ki分别为21.5 nM和303.3 nM。Phase 2。 |
-
iScience, 2024, 27(10):110862
-
J Pers Med, 2022, 12(2)258
-
Hum Cell, 2022, 10.1007/s13577-022-00671-y
|

|
| S7358 |
Poziotinib (NOV120101, HM781-36B)
|
Poziotinib是一种不可逆的泛-HER抑制剂,对HER1,HER2,和HER4的IC50分别为3.2 nM,5.3 nM和23.5 nM。Poziotinib可诱导凋亡和G1细胞周期阻滞。Phase 2。 |
-
Autophagy, 2026, 1-27.
-
Clin Cancer Res, 2025, 31(14):3002-3018
-
Nat Biomed Eng, 2024, 10.1038/s41551-024-01273-9
|
|
| S1173 |
WZ4002
|
WZ4002是一种新型的,突变选择性EGFR抑制剂,作用于EGFR(L858R)/(T790M),在BaF3细胞系中IC50为2 nM/8 nM;对ERBB2磷酸化(T798I)没有抑制作用。 |
-
J Extracell Vesicles, 2024, 13(7):e12494
-
Cancer Metab, 2023, 11(1):20
-
Cancer Gene Ther, 2022, 29(12):1878-1894
|

|
| S8362 |
Tucatinib (Irbinitinib, ONT-380, Arry-380)
|
Tucatinib (Irbinitinib, ONT-380, ARRY-380) 是一种具有口服活性、可逆的、ATP竞争性的的ErbB2(HER2)小分子抑制剂。在细胞实验中,ARRY-380对ErbB-2和p95 HER2的IC50s分别为8 nM和7 nM,对HER2的选择性是对EGFR的500倍。具有潜在的抗肿瘤活性。 |
-
Biochem Pharmacol, 2026, 250(Pt 1):117962
-
Cancer Cell, 2025, 43(4):776-796.e14
-
Cancer Discov, 2025, 10.1158/2159-8290.CD-25-0605
|
|
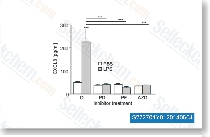
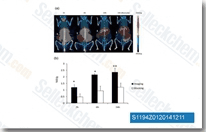
| S1194 |
CUDC-101
|
CUDC-101是一种有效的,多靶点抑制剂,作用于HDAC,EGFR和HER2,IC50分别为4.4 nM, 2.4 nM,和15.7 nM,且抑制I/II型HDACs,但是对III型, Sir-type HDACs没有抑制作用。Phase 1。 |
-
Oncotarget, 5 Jul 2017, 80109-80123
-
ACS Medicinal Chemistry Letters, 25 Jul 2013, 858-862
-
J Am Heart Assoc, 2025, 14(1):e037400
|
|
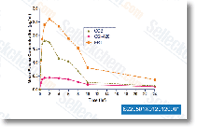
| S2205 |
DesMethyl Erlotinib (OSI-420) HCl
|
DesMethyl Erlotinib (OSI-420) HCl是Erlotinib的活性代谢产物(是一种EGFR抑制剂,IC50为2 nM)。 |
-
Drug Metab Dispos, 2025, 53(5):100069
-
J Pharm Biomed Anal, 2023, 236:115716
-
Front Oncol, 2022, 12:939118
|
|
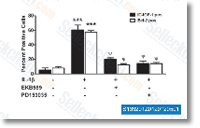
| S1079 |
PD153035 HCl
|
PD153035 HCl (SU-5271 HCl, AG1517 HCl, ZM 252868 HCl)是一种有效的,特异性EGFR抑制剂,无细胞试验中Ki和IC50分别为5.2 pM和29 pM;对PGDFR, FGFR, CSF-1, InsR和Src几乎无作用。 |
-
Journal of Experimental & Clinical Cancer Research, July 29, 2020, 145
-
Molecular Cancer Therapeutics, January 2015, 298-306
-
Nature Communications, December 9, 2025, 1
|
|
| S2185 |
Allitinib tosylate
|
Allitinib (AST-1306, AST-6) 是一种新型,不可逆的EGFR和ErbB2抑制剂,IC50分别为0.5 nM和3 nM,对突变型EGFR T790M/L858R也有效,作用于过表达ErbB2的细胞更有效,作用于ErbB家族比作用于其他激酶选择性高3000倍。 |
-
Mol Oncol, 2025, 10.1002/1878-0261.70096
-
Hum Cell, 2024, 10.1007/s13577-024-01054-1
-
Research Square, 2023, Version 1
|

|
| S1486 |
AEE788 (NVP-AEE788)
|
AEE788 (NVP-AEE788)是一种有效的EGFR和HER2/ErbB2抑制剂,IC50分别为2 nM和6 nM,对VEGFR2/KDR, c-Abl, c-Src,和Flt-1作用效果稍弱,对Ins-R, IGF-1R, PKCα和CDK1没有抑制作用。Phase 1/2。 |
-
Cell Rep Med, 2022, 3(1):100492
-
Genome Med, 2020, 18;12(1):17
-
Oncotarget, 2019, 10(68):7185-7197
|

|
| S6546 |
PD153035
|
PD153035是一种特定的、有效的EGFR抑制剂,Ki值为5.2 pM。 |
-
Biomed Pharmacother, 2023, 158:114176
-
Cell Death Discov, 2023, 10.1038/s41420-023-01701-w
-
Sci Signal, 2022, 15(728):eabj6915
|
|
| S1056 |
AC480 (BMS-599626)
|
AC480 (BMS-599626)是一种选择性的,高效效的HER1和HER2抑制剂,IC50分别为20 nM和30 nM。比对HER4效果强8倍左右,而比对VEGFR2, c-Kit, Lck, MET等的作用强100倍以上。Phase 1。 |
-
Communications Biology, 2025, Article number: 1350
-
Int J Mol Sci, 2022, 23(22)14023
-
Genome Med, 2020, 18;12(1):17
|
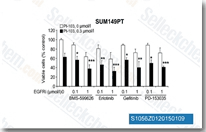
|
| S1392 |
Pelitinib (EKB-569)
|
Pelitinib (EKB-569) 是一种有效的,不可逆 EGFR 抑制剂,IC50为38.5 nM。Pelitinib (EKB-569) 还轻微抑制 Src、MEK/ERK 和 ErbB2 ,对应的IC50值分别为282 nM、800 nM和1255 nM。Phase2。 |
-
Cancer Cell, 2022, S1535-6108(22)00312-9
-
Cancer Discov, 2022, 12(5):1378-1395
-
Cell Oncol (Dordr), 2022, 45(4):601-619
|
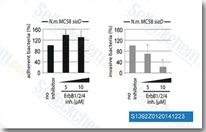
|
| S2922 |
Icotinib (BPI-2009H)
|
Icotinib (BPI-2009H)是一种有效的,特异性EGFR抑制剂,IC50为5 nM,包括EGFR, EGFR(L858R), EGFR(L861Q), EGFR(T790M)和EGFR(T790M, L858R)。Phase 4。 |
-
iScience, 2024, 27(10):110862
-
Am J Cancer Res, 2024, 14(1):33-51
-
Cell Rep Med, 2022, 3(1):100492
|
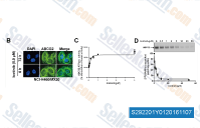
|
| S2867 |
WHI-P154
|
WHI-P154是一种有效的JAK3抑制剂,IC50为1.8 μM,对JAK1和JAK2没有抑制活性,也抑制EGFR, Src, Abl, VEGFR和MAPK,抑制Stat3而非Stat5磷酸化。 |
-
Nat Commun, 2023, 14(1):6908
-
Int J Mol Sci, 2023, 10.3390/ijms241813863
-
Int J Mol Sci, 2023, 24(18)13863
|
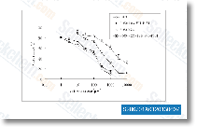
|
| E0822 |
AST-1306
|
AST-1306 (Allitinib)是一种口服活性和不可逆的EGFR、ErbB2和ErbB4抑制剂,IC50s分别为0.5,3和0.8 nM。 |
-
Mol Oncol, 2025, 10.1002/1878-0261.70096
-
Molecular Oncology, 2025, 3205-3222
-
Human Cell, 2024, 1170-1183
|
|
| S8724 |
Lazertinib (YH25448)
|
Lazertinib (YH25448,GNS-1480)是一种有效的、对突变型具有高选择性的、不可逆的EGFR抑制剂,对Del19/T790M、L858R/T790M、Del19、L85R和野生型EGFR的IC50分别为1.7 nM, 2 nM, 5 nM, 20.6 nM和76 nM。对ErbB2和ErbB4的IC50值更高。 |
-
Cell Rep Med, 2025, 6(2):101929
-
Cells, 2025, 14(17)1386
-
Front Oncol, 2025, 15:1533059
|
|
| S2755 |
Varlitinib
|
Varlitinib (ARRY334543) 是一种有效的,选择性ErbB1(EGFR)和ErbB2(HER2)抑制剂,IC50分别为7 nM和2 nM。Phase 2。 |
-
J Transl Med, 2023, 21(1):89
-
Cell Rep Med, 2022, 3(1):100492
-
Pharmaceutics, 2019, 11(11)
|

|
| S0711 |
Canertinib dihydrochloride
|
Canertinib (CI-1033, PD-183805, compound 18) dihydrochloride 是一种有效且不可逆的 epidermal growth factor receptor (EGFR) tyrosine kinase 的抑制剂。Canertinib dihydrochloride 可抑制细胞 EGFR 和 ErbB2 自磷酸化,IC50值分别为 7.4 nM和 9 nM。 |
-
Front Microbiol, 2022, 13:968872
-
Acta Physiol (Oxf), 2021, 10.1111/apha.13661
-
Sci Rep, 2017,
|
|
| S7926 |
Lifirafenib (BGB-283)
|
Lifirafenib (BGB-283, Beigene-283) 有效地抑制RAF激酶家族和EGFR活性,在生化实验检测中,其对重组BRAF(V600E)激酶区域、EGFR和EGFR(T790M/L858R)突变体的IC50分别为23、29、495 nM。 |
-
Cancer Research Communications, 2025, nan
-
Sci Adv, 2023, 9(35):eade7486
-
Mol Cancer Ther, 2022, MCT-22-0004
|
|
| S8312 |
NSC228155
|
NSC228155是EGFR的激活剂,与sEGFR的二聚体化功能域II结合、调节EGFR的酪氨酸磷酸化。 |
-
JCI Insight, 2025, e173700
-
Int J Oncol, 2025, 66(3)20
-
Adv Sci (Weinh), 2023, 10(24):e2207631
|
|
| S7971 |
Zorifertinib (AZD3759)
|
Zorifertinib (AZD3759)是一种有效的,具有口服活性的,CNS-渗透性EGFR抑制剂,对EGFR (WT),EGFR (L858R),和 EGFR (exon 19Del)的IC50分别为0.3 nM,0.2 nM,和 0.2 nM。Phase 1。
|
-
Frontiers in Immunology, June 6, 2025, 1606125
-
Drug Metabolism and Disposition, April 2019, 393-404
-
Drug Des Devel Ther, 2024, 18:1175-1188
|
|
| S7039 |
PD168393
|
PD168393是一种不可逆的EGFR抑制剂,IC50为0.70 nM,不可逆烷基化Cys-773;抑制insulin, PDGFR, FGFR和PKC活性。 |
-
Elife, 2024, 13e85914
-
FEBS Open Bio, 2023, 10.1002/2211-5463.13653
-
J Neurosci Res, 2021, 10.1002/jnr.24909
|
|
| S7298 |
AZ5104
|
AZ5104,是AZD-9291的脱甲基代谢物,是一种有效的EGFR抑制剂,作用于EGFR (L858R/T790M),EGFR (L858R),EGFR (L861Q),和EGFR (野生型),IC50分别为<1 nM,6 nM,1 nM,和25 nM。Phase 1。 |
-
Genes & Diseases, 2023, 2241-2244
-
J Pharm Biomed Anal, 2020, 177:112871
-
Clin Cancer Res, 2019, 25(8):2575-2587
|
|
| S8294 |
Olmutinib (BI 1482694)
|
Olmutinib (BI 1482694) 是一种新型的EGFR突变特效性酪氨酸激酶抑制剂、布鲁顿氏酪氨酸激酶抑制剂。 |
-
iScience, 2024, 27(10):110862
-
Br J Pharmacol, 2022, 179(11):2754-2770
-
Ther Adv Med Oncol, 2022, 14:17588359221079125
|
|
| S7824 |
Nazartinib (EGF816)
|
Nazartinib (EGF816, NVS-816)是共价的、不可逆的、对突变型EGFR具有选择性的抑制剂,对突变型EGFR(L858R, ex19del)和T790M具有纳摩尔级别的抑制作用,相对于野生型EGFR,其对突变型EGFR更具特异性。 |
-
Translational Oncology, March 2019, 705-713
-
Cancer Cell, July 11, 2022, 754-767.e6
-
Cancer Cell, 2022, 40(7):754-767.e6
|
|
| S8852 |
Pyrotinib dimaleate
|
Pyrotinib dimaleate是一种有效的、具有选择性的、不可逆的 EGFR 和 HER2 双重酪氨酸激酶抑制剂,其对应的IC50值分别为0.013 μM和0.038 μM。 |
-
Cancer Research, 2026, nan
-
Cancer Research, 2025 Oct 15, 3983-3998
-
Cancer Research, 2025, 3983-3998
|
|
| S7000 |
AP26113-analog (ALK-IN-1)
|
AP26113-analog (ALK-IN-1) 是AP26113的类似物,是有效的、选择性的ALK抑制剂,同时也是EGFR的抑制剂。 |
-
ChemMedChem, 2017, 1219-1230
-
Clin Cancer Res, 2015, 21(1):166-74
-
Clin Cancer Res, 2014, 20(22):5686-96
|

|
| S2784 |
TAK-285
|
TAK-285是一种新型,HER2和EGFR(HER1)双重抑制剂,IC50分别为17 nM和23 nM,作用于HER1/2比作用于HER4选择性高10倍以上,对MEK1/5, c-Met, Aurora B, Lck, CSK等作用效果稍弱。Phase 1。 |
-
Oncotarget, 2020, 11(46):4224-4242
-
Oncotarget, 2020, 4224-4242
-
Mol Pharmacol, 2019, 95(5):528-536
|
|
| S7557 |
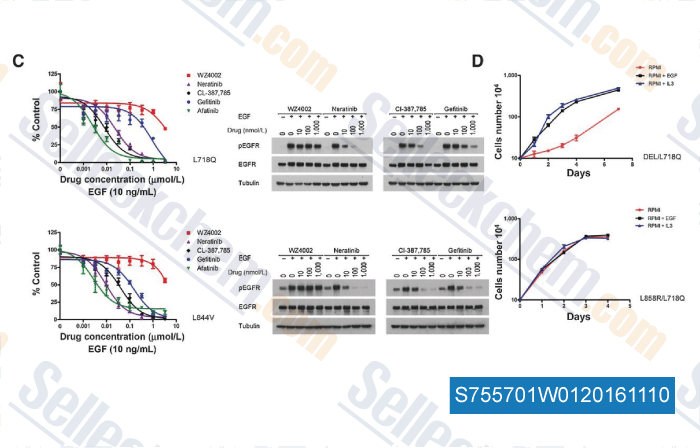
CL-387785 (EKI-785)
|
CL-387785 (EKI-785, WAY-EKI 785)是一种不可逆的选择性EGFR抑制剂,IC50为370 pM。
|
-
iScience, 2025, 28(2):111708
-
Stem Cell Reports, 2023, 10.1016/j.stemcr.2023.03.011
-
Nature, 2017, 550(7675):270-274
|
|
| S6813 |
Mobocertinib (TAK788)
|
Mobocertinib (TAK788, AP32788) 是一种研究用的TKI,是有效的、选择性的 epidermal growth factor receptor (EGFR) 和 HER2 突变体的临床前抑制剂。Mobocertinib (TAK788) 是一种抗肿瘤药。 |
-
Int J Mol Sci, 2024, 25(7)3992
-
J Thorac Oncol, 2023, S1556-0864(23)00797-9
-
Genes & Diseases, 2023, 2241-2244
|
|
| S3759 |
Norcantharidin
|
Norcantharidin (Endothall anhydride) is a synthetic anticancer compound which is a dual inhibitor for c-Met and EGFR in human colon cancers. |
-
Frontiers in Pharmacology, November 3, 2022, 1019478
-
Frontiers in Pharmacology, November 02, 2022, 1019478
-
Molecular Medicine Reports, March 8, 2024, 71
|
|
| S8814 |
zipalertinib (TAS6417)
|
zipalertinib (TAS6417)是一种新型的EGFR抑制剂,靶向EGFR突变体,对野生型EGFR抑制效力较低,IC50范围为1.1 ± 0.1到8.0 ± 1.1 nmol/L。 |
-
iScience, 2024, 108839
-
iScience, 2024, 27(2):108839
-
STAR Protocols, 2024, 102987
|
|
| S2554 |
Daphnetin
|
Daphnetin是天然香豆素衍生物,是一种蛋白激酶抑制剂,抑制EGFR, PKA和PKC,IC50分别为7.67 μM, 9.33 μM和25.01 μM,且具有抗炎和抗氧化剂活动性。 |
-
Sci Rep, 2021, 11(1):11491
-
Acta Neuropathol, 2020, 10.1007/s00401-020-02226-7
-
Int Immunopharmacol, 2019, 72:195-203
|
|
| S0360 |
EGFR Inhibitor
|
EGFR inhibitor 是一种可透过细胞的4,6-二取代嘧啶化合物,是一种具有高度选择性的 EGFR kinase 的抑制剂,其IC50值为21 nM。EGFR inhibitor 可直接使 microtubules 解聚,可作为研究EGFR途径和微管动力学的化学探针。 |
-
Cell Rep, 2022, 40-8:111223
-
Frontiers in Pharmacology, 2022, 804327
-
Cancers (Basel), 2022, 14(19)4910
|
|
| S8817 |
Almonertinib (HS-10296)
|
Almonertinib (HS-10296)是一种EGFR激活突变和对EGFR T790M具有耐受性突变的抑制剂,其对野生型EGFR仅具有有限活性。 |
-
Cells, 2025, 14(17)1386
-
iScience, 2024, 27(10):110862
-
Biomedicines, 2024, 12(7)1412
|
|
| S1179 |
WZ8040
|
WZ8040是一种新型的,突变选择性的,不可逆的EGFRT790M抑制剂,对ERBB2磷酸化(T798I)没有抑制作用。 |
-
J Biol Chem, 2025, 301(8):110454
-
Biochemistry, 2018, 57(8):1369-1379
-
Nat Commun, 2016, 7:10690
|

|
| E4819 |
(E/Z)-Afatinib
|
(E/Z)-Afatinib (Gilotrif, BIBW2992, Boehringer Ingelheim)是第二代酪氨酸激酶抑制剂(TKI),对EGFR和HER2具有不可逆抑制作用,对野生型EGFR、EGFR-L858R、HER2、ErbB4的IC50分别为0.5 nM、0.4 nM、14 nM和1 nM。 |
-
Cell Death & Disease, 2021, 728
-
Gynecologic Oncology, 2019 Apr, 158-164
-
Bioscience Reports, 2018, BSR20181964
|
|
| S6868 |
Furmonertinib Mesylate (AST2818, Alflutinib)
|
Alflutinib (Furmonertinib) mesylate是第三代 epidermal growth factor receptor (EGFR) 的抑制剂,可抑制EGFR敏感突变和T790M突变。Alflutinib (AST2818) 主要由CYP3A4代谢,也是有效的 CYP3A4 的诱导剂,EC50值为0.25 μM。 |
-
Cells, 2025, 14(17)1386
-
Biomedicines, 2024, 12(7)1412
-
Int J Mol Sci, 2023, 24(18)13972
|
|
| S2406 |
Chrysophanic Acid
|
Chrysophanic Acid (Chrysophanol) 是Dianella longifolia中的一种天然蒽醌,是一种EGFR/mTOR通道抑制剂。 |
-
Cell Proliferation, June 29, 2020, e12871
-
International Journal of Oncology, February 11, 2025, 20
-
Int J Oncol, 2025, 66(3)20
|

|
| S9786 |
Tuxobertinib (BDTX-189)
|
Tuxobertinib (BDTX-189)是一种有效的 EGFR 和 HER2 致癌突变的变构性抑制剂,对EGFR、HER2、BLK 和 RIPK2的Kd值分别为0.2 nM、0.76 nM、13 nM和1.2 nM。BDTX-189 具有抗癌活性。 |
-
Molecular Pharmacology, 2024 Jan 10, 97-103
-
Chem Biol Interact, 2024, 395:111033
-
J Transl Med, 2023, 21(1):89
|
|
| S4667 |
Lidocaine hydrochloride
|
Lidocaine hydrochloride (Lidothesin, Lignocaine, Xyloneural)是一种 epidermal growth factor receptor (EGFR) 的抑制剂,对人舌癌细胞具有抗增殖作用。Lidocaine hydrochloride 是一种局部麻醉剂及心脏抑制剂,被用作抗心律失常剂。 |
-
Cancer Research, July 1, 2021, 3580-3592
-
J Immunother Cancer, 2024, 12(11)e009805
-
Exp Ther Med, 2021, 21(5):424
|
|
| S8036 |
Butein
|
Butein,一种从漆树中分离的植物多酚,可以抑制酪氨酸激酶,NF-κB,STAT3和EGFR活化。 |
-
Scientific Reports, 2022, 20644
-
American Journal of Cancer Research, 2020, 3721-3736
-
Am J Cancer Res, 2020, 10(11):3721-3736
|
|
| S8242 |
EAI045
|
EAI045是一种靶向特定耐药性的EGFR突变体的变构抑制剂。对野生型受体无作用。 |
-
bioRxiv, August 22, 2021, nan
-
EJNMMI Res, 2024, 14(1):19
-
Pharmazie, 2018, 73(11):630-634
|
|
| E4886 |
HS-10296 hydrochloride
|
HS-10296 hydrochloride(Almonertinib (hydrochloride), HS-10296 HCl) 是一种不可逆的第三代 EGFR 酪氨酸激酶 抑制剂,对 EGFR 敏感突变、T790M 抗性突变具有高选择性。它对 T790M、T790M/L858R 和 T790M/Del19 具有抑制活性,IC50 分别为 0.37 nM、0.29 nM 和 0.21 nM。它还具有抗肿瘤活性。 |
-
Cells, 2025, 1386
-
Cancer Medicine, 2025, e70827
-
Advanced Science, 2024, e2405130
|
|
| S8741 |
Avitinib (Abivertinib)
|
Avitinib (Abivertinib)是不可逆的EGFR抑制剂,对于突变型EGFR具有选择性,对EGFR L858R/T790M双突变体的IC50为0.18 nM,是对野生型EGFR效力的近43倍(IC50=7.68 nM for WT EGFR)。Avitinib具有抗肿瘤活性和耐受性毒性。 |
-
iScience, 2024, 27(10):110862
-
Microsyst Nanoeng, 2023, 9:57
-
, 2022, 10.21203/rs.3.rs-2146794/v1
|
|
| S1170 |
WZ3146
|
WZ3146是一种突变选择性的,不可逆EGFR(L858R)和EGFR(E746_A750)抑制剂,IC50分别为2 nM和2 nM;对ERBB2磷酸化(T798I)没有抑制作用。 |
-
Sci Rep, 2024, 14(1):22895
-
Aging (Albany NY), 2020, 12(19):19022-19044
|

|
| S2895 |
Tyrphostin 9
|
Tyrphostin 9 (SF 6847, RG-50872) 是第一个EGFR抑制剂,IC50为460 μM,作用于PDGFR更有效,IC50为0.5μM。 |
-
PLoS One, June 2, 2020, e0233993
-
Cell Rep, 2022, 38(10):110475
|
|
| S7206 |
CNX-2006
|
CNX-2006是一种新型,不可逆,突变选择性的EGFR抑制剂,IC50为<20 nM,对野生型EGFR有微弱的抑制作用。 |
-
Nat Nanotechnol, 2021, 10.1038/s41565-021-00872-w
-
Cancer Science, 2016, 461-468
-
Cancer Sci, 2016, 107(4):461-8
|

|
| E5906 |
Sunvozertinib
|
Sunvozertinib (DZD9008) 是一种口服、强效且选择性的 ErbBs(EGFR 或 Her2) 抑制剂,特别是 ErbBs 突变形式。它还可抑制 EGFR 外显子 20 NPH 插入、EGFR 外显子 20 ASV 插入、EGFR L858R 和 T790M 突变、Her2 外显子 20 YVMA 和 EGFR WT A431,IC50 值分别为 20.4 nM、20.4 nM、1.1 nM、7.5 nM 和 80.4 nM。 |
-
Journal of Thoracic Oncology, 2024, 71-79
-
bioRxiv , 2023, 10.1101/2023.07.27.550902
|
|
| S6541 |
MTX-211
|
MTX-211是PI3K和EGFR的双重抑制剂。 |
-
Int J Mol Sci, 2024, 25(10)5160
-
Int J Mol Sci, 2023, 24(8)7608
|
|
| S8412 |
Naquotinib(ASP8273)
|
Naquotinib (ASP8273)是一种具有口服活性的、不可逆的、对突变体具有选择性的epidermal growth factor receptor (EGFR)抑制剂,具有潜在的抗肿瘤活性。 |
-
ACS Infectious Diseases, 2019, 1764-1771
-
Expert Opin Emerg Drugs, 2018, 10.1080/14728214.2018.1558203
Share on FacebookShare on TwitterShare on Google+
|
|
| S2752 |
HER2-Inhibitor-1
|
HER2-Inhibitor-1是ARRY-380的类似物。ARRY-380是一种有效的,选择性的HER2抑制剂,IC50为8 nM,等效作用于截短的p95-HER2,作用于HER2比作用于EGFR选择性高500倍。 |
-
Communications Biology, 2021, 762
-
Commun Biol, 2021, 4(1):762
|

|
| S8009 |
AG-18
|
AG-18 (RG-50810, Tyrphostin A23, TX 825)抑制EGFR,IC50为35 μM。 |
-
Reproduction, 2016, 151(2):179-85
|
|
| S6899 |
Licochalcone D
|
Licochalcone D (Lico D, LCD, LD) 是一种从中草药甘草 Glycyrrhiza inflata 中分离出的类黄酮,具有抗氧化、抗炎和抗癌的特性。Licochalcone D 可抑制LPS信号通路中NF-κB p65的磷酸化。Licochalcone D 可抑制 JAK2、EGFR 和 Met (c-Met) 活性,并诱导ROS依赖性的凋亡。Licochalcone D 还可诱导 caspases 的活化和 poly (ADP-ribose) polymerase (PARP) 的裂解。 |
-
Frontiers in Pharmacology, September 18, 2025, 16:1573882
|
|
| S6525 |
AG 555
|
AG-555 (Tyrphostin B46)是酪氨酸激酶抑制剂,可与拓扑异构酶I直接相互作用,因而阻止DNA松弛。它抑制EGFR,IC50为0.7 μM。 |
-
Tesi Doctoral, Universitat Autònoma de Barcelona, 2017, 99
|
|
| E1176 |
BLU-945
|
BLU-945是一种强效、高选择性、可逆、口服活性表皮生长因子受体酪氨酸激酶(EGFR)抑制剂(TKIs)。BLU-945可用于肺癌包括非小细胞肺癌(NSCLC)的研究。 |
-
Eur J Pharm Sci, 2025, S0928-0987(25)00335-5
|
|
| S9711 |
CH7233163
|
CH7233163 是一种 EGFR-tyrosine kinase 的非共价ATP竞争性抑制剂,对有EGFR-Del19/T790M/C797S的肿瘤具有抗肿瘤活性。 |
-
bioRxiv , 2023, 10.1101/2023.07.27.550902
|
|
| E1791 |
Zongertinib (BI 1810631)
|
Zongertinib (BI 1810631, BI 764532)是一种有效的选择性人HER2和EGFR酪氨酸激酶抑制剂,共价结合野生型和突变的HER2受体,包括外显子20插入突变,同时保留EGFR信号;口服Zongertinib治疗HER2异常阳性的实体瘤有效且耐受。 |
-
Cancer Discov, 2025, 10.1158/2159-8290.CD-25-0605
|
|
| S6698 |
TQB3804
|
TQB3804是一种选择性有效的 EGFR 激酶抑制剂,对于 EGFRd746-750/T790M/C797S、EGFRL858R/T790M/C797S、EGFRd746-750/T790M、EGFRL858R/T790M 和 EGFRWT的IC50值分别为0.46 nM、0.13 nM、0.26 nM、0.19 nM 和 1.07 nM。TQB3804 (EGFR-IN-7) 也具有抗肿瘤活性。 |
-
Signal Transduction and Targeted Therapy, 2025, 389
-
Signal Transduct Target Ther, 2025, 10(1):389
|
|
| S8921 |
BI-4020
|
BI-4020 是一种口服活性的 EGFR 的非共价抑制剂,其对 EGFRdel19 T790M C797S 的在BaF3细胞中的生物标志物效力和抗增殖效力对应的IC50分别为0.6 nM和0.2 nM。 |
-
Biomedicines, 2024, 12(7)1412
-
Biomedicines, 2024, 1412
|
|
| E0368 |
(S)-Sunvozertinib ((S)-DZD9008)
|
(S)-Sunvozertinib ((S)-DZD9008) 是一种口服、有效、不可逆、野生型选择性 EGFR 抑制剂,可抑制 EGFR 或 HER2 Exon20ins 和其他突变体,对突变型 EGFR 的 IC50 范围为 0.4 nM 至 2.1 nM。 |
-
J Thorac Oncol, 2023, S1556-0864(23)00797-9
|
|
| E1919 |
Befotertinib mesylate
|
Befotertinib mesylate (D-0316 mesylate) 是第三代EGFR酪氨酸激酶抑制剂(TKI),可用于EGFR T790M阳性非小细胞肺癌(NSCLC)的研究。 |
-
Cells, 2025, 14(17)1386
|
|
| E2399 |
Falnidamol
|
Falnidamol (BIBX1382)是一种选择性的表皮生长因子受体(epidermal growth factor receptor, EGFR)抑制剂,通过抑制丝裂原活化蛋白激酶(mitogen-activated protein kinases, MAPKs)信号传导,在体外和体内对人头颈部鳞状细胞癌发挥抗癌作用。 |
-
Free Radical Biology and Medicine, 2022, 106-124
|
|
| E4675 |
Rezivertinib
|
Rezivertinib(BPI-7711) 是一种有效的第三代 EGFR 酪氨酸激酶 抑制剂,可同时靶向 EGFR 敏感突变和 EGFR T790M 突变。此外,它具有出色的中枢神经系统渗透性,并具有抗肿瘤活性。 |
-
Cells, 2025, 14(17)1386
-
Cells, 2025, 1386
|
|
| S9414 |
Cyasterone
|
Cyasterone is the main phytoecdysteroid component found in Cyathula capitata. It is a natural EGFR inhibitor and maybe a promising anti-cancer agent. |
-
Chin Med, 2023, 18(1):136
|
|
| S6897 |
Epertinib hydrochloride
|
Epertinib hydrochloride (S-222611 hydrochloride) 是有一种效的、口服活性的、可逆的、选择性的 tyrosine kinase 抑制剂,针对 EGFR、HER2 和 HER4,对应的IC50值分别为1.48 nM、7.15 nM和2.49 nM。Epertinib hydrochloride (S-222611 hydrochloride) 具有抗肿瘤活性。 |
-
Biomedicine & Pharmacotherapy, 2024, 117542
|
|
| E4904 |
Dacomitinib hydrate
|
Dacomitinib hydrate(PF-00299804, PF-804)是一种口服给药、高度选择性的不可逆抑制剂,靶向作用于泛ERBB受体,对EGFR的IC50为6.0 nM,对ERBB2的IC50为45.7 nM,对ERBB4的IC50为73.7 nM。此外,它还表现出抗肿瘤活性。 |
-
McGill University, Montreal, 2021,
|
|
| E2980 |
4-AMino-1-phenylpyrazolo[3,4-d]pyriMidine
|
4-AMino-1-phenylpyrazole[3,4-d]pyriMidine(PP 3)是一种 EGFR 酪氨酸激酶抑制剂,IC50 为 2.7 μM。 它是 Src 激酶抑制剂 PP 2 的阴性对照。 |
-
Molecular Biology of the Cell, March 01, 2021, 376-390
-
Molecular Biology of the Cell, 2021, 376-390
|
|
| E4449 |
AG 825
|
AG-825 (Tyrphostin AG-825) 是一种有效的选择性 ErbB2 抑制剂,IC50 值为 0.15 µM。 它还抑制 PDGFR 自磷酸化,IC50 值为 40 µM。 它具有抗癌活性并显着诱导癌细胞凋亡。 |
|
|
| E1749 |
limertinib
|
Limertinib (ASK120067) 是一种不可逆的第三代 EGFRT790M 抑制剂,IC50 为 0.3 nM,选择性优于 EGFRWT< /b> IC50 为 6.0 nM。 它在非小细胞肺癌 (NSCLC) 肿瘤细胞中表现出有效的抗增殖活性。 |
|
|
| S6805 |
Tyrphostin AG-528
|
Tyrphostin AG-528 (Tyrphostin B66) 是一种 epidermal growth factor receptors (EGFR) 和 ErbB2/HER2 的有效抑制剂,其IC50值分别为4.9 μM和2.1 μM。Tyrphostin AG-528 具有抗癌活性。 |
|
|
| E5912 |
Asandeutertinib
|
Asandeutertinib (Osimertinib-d3, AZD-9291-d3) 是一种口服、ATP 竞争性、不可逆、氘化的第三代表皮生长因子受体酪氨酸激酶抑制剂 (EGFR TKI),也是osimertinib 的氘稳定化衍生物,具有治疗晚期 EGFR 突变 NSCLC 的潜力。 |
|
|
| S6509 |
AG 494
|
AG-494是EGFR受体自身磷酸化的抑制剂,IC50为1.2 μM。它抑制依赖于EGF的细胞生长,IC50为6 μM。 |
|
|
| S0736 |
AG 556
|
AG-556是一种有效的EGFR酪氨酸抑制剂,IC50为5µM,可抑制EGF诱导的HER14细胞生长。 |
|
|
| S6523 |
RG14620
|
RG14620 (Tyrphostin RG14620)是一种EGFR抑制剂,可直接抑制ABCG2/BCRP的传送功能。 |
|
|
| S6530 |
EBE-A22
|
EBE-A22是PD 153035的衍生物。PD 153035可抑制ErbB-1磷酸化,而EBE-A22无此活性。 |
|
|
| E1877 |
Pebezertinib (BLU-451, LNG-451)
|
Pebezertinib 是一种中枢神经系统(CNS)渗透性,保留野生型EGFR功能,针对EGFR exon 20 insertion (Ex20ins)的共价抑制剂,IC50为7-78 nM,具有强大的抗肿瘤功效。 |
|
|
| S0070 |
Gefitinib-based PROTAC 3
|
Gefitinib-based PROTAC 3 通过连接器将EGFR结合元件与VHL配体偶联,可诱导 EGFR 及其突变体的降解,在HCC827(第19外显子)和H3255(L858R)中的DC50值分别为11.7 nM和22.3 nM。 |
|
|
| S0324 |
Tyrphostin AG30 (AG30)
|
Tyrphostin AG30 (AG30)是一种有效的选择性EGFR酪氨酸激酶抑制剂,选择性抑制c-ErbB诱导的自我更新,并能够抑制c-ErbB在原代成红细胞中对STAT5的激活。 |
|
|
| E0344 |
ErbB2 inhibitor
|
ErbB-2 Inhibitor是一种EGFR、ErbB2和ErbB4抑制剂,IC50分别为0.017 μM, 0.08 μM, 1.91 μM。 |
|
|
| E4597 |
Avitinib maleate
|
Avitinib maleate(Abivertinib maleate, AC0010 maleate, AC0010MA) 是第三代、不可逆且口服有效的 EGFR 选择性抑制剂。其 IC50 值分别为 0.18 nM、0.18 nM、7.68 nM,针对 EGFR L858R、EGFR T790M 和野生型 EGFR。Avitinib maleate还可作为 BTK 抑制剂,诱导套细胞淋巴瘤中的 BTK 凋亡。 |
|
|
| E4664 |
Osimertinib dimesylate
|
Osimertinib dimesylate (Mereletinib dimesylate, AZD-9291 dimesylate) 是一种强效、突变选择性、不可逆的 EGFR 激酶活性抑制剂,对 EGFRL858R 和 EGFRL858R/T790M 的 IC50 分别为 12 nM和 1 nM。它在异种移植和转基因 T790M EGFR 突变模型中表现出抗肿瘤活性,支持其在非小细胞肺癌 (NSCLC) 研究中的应用。 |
|
|
| S3879 |
Kaempferide
|
Kaempferide (4'-Methylkaempferol, 4'-O-Methylkaempferol, Kaempferol 4'-methyl ether) 是一种从山柰根中提取的天然产物,具有多种生物活性,包括抗癌、抗炎、抗氧化、抗菌抗病毒等活性。 |
|
|
| S2733 |
AV-412 free base
|
AV-412 free base (MP-412 free base)是一种EGFR抑制剂,对EGFR,EGFRL858R,EGFRT790M,EGFRL858R/T790M和ErbB2的IC50分别为0.75,0.51,0.79,2.30和19 nM。 |
|
|
| S8920 |
(Rac)-JBJ-04-125-02
|
(Rac)-JBJ-04-125-02 (JBJ-04-125-02 racemate) 是一种 Epidermal growth factor receptor (EGFR) 的突变体选择性的变构抑制剂,具有潜在的抗癌活性。 |
|
|
| S6881 |
JND3229
|
JND3229 是一种有效且可逆的EGFRC797S抑制剂,IC50为5.8 nM。JND3229还有效抑制EGFRL858R/T790M和EGFRWT,IC50值分别为30.5 nM 和6.8 nM。 |
|
|
| S1054 |
AG99
|
AG99 (Tyrphostin 46,Tyrphostin A46,Tyrphostin B40)是一种有效、选择性的EGFR抑制剂。 |
|
|
| S8584 |
Theliatinib (HMPL-309)
|
Theliatinib (HMPL-309)是一种高效的EGFR抑制剂,对EGFR的Ki值为0.05 nM,对EGFR和EGFR T790M/L858R突变体的IC50值分别为3 nM和22 nM。它对EGFR的选择性是对其他72种激酶的50倍以上。 |
|
|
| E6000 |
BIEGi-1
|
BIEGi-1 是一种强效且选择性的EGFR二元抑制剂,可有效阻断 EGFR 催化的核苷酸交换并破坏 EGFR-Rheb 相互作用。它能够强效抑制 EGFR 突变癌细胞中的 EGFR 激酶活性和 mTORC1 信号传导。BIEGi-1 表现出显著的抗增殖作用,对 PC9 的 IC50 为 17 nM,对 HCC827 的 IC50 为 20 nM。 |
|
|
| S0151 |
AG-1557
|
AG-1557 是一种特异性的 epidermal growth factor receptor (EGFR) 酪氨酸激酶的ATP竞争性抑制剂。 |
|
|
| S2115 |
RG 13022
|
RG 13022 (Tyrphostin RG13022)抑制免疫沉淀反应中EGF receptor的自磷酸化反应,IC50为4 µM。 |
|
|
| E4884 |
Icotinib Hydrochloride
|
Icotinib hydrochloride (BPI-2009H) 是一种高效、选择性 表皮生长因子受体酪氨酸激酶抑制剂 (EGFR-TKI),IC50 为 5 nM,用于治疗非小细胞肺癌 (NSCLC)。 |
|
|
| E6528New |
BBT-176
|
BBT-176 是一种首创的可逆性第四代EGFR TKI,对奥希替尼耐药突变具有强效活性,在 Ba/F3 细胞中 IC50 分别为 49 nM (Del19/T790M/C797S)、202 nM (L858R/T790M/C797S)、42 nM (Del19/C797S) 和 183 nM (L858R/C797S)。 |
|
|
| E1478 |
BDTX-1535
|
BDTX-1535 (EGFR-IN-76) 是一种口服、高效、选择性和不可逆的 EGFR 抑制剂,可以穿透血脑屏障,具有治疗胶质母细胞瘤或非小细胞瘤的潜力 细胞肺癌。 |
|
|
| E4997 |
Sevabertinib (BAY 2927088)
|
BAY 2927088(Sevabertinib)是首个非共价、强效、口服、选择性、可逆性 酪氨酸激酶 抑制剂,在临床前模型中可强效抑制突变型 HER2 和突变型 EGFR。该药物靶向非小细胞肺癌 (NSCLC) 患者的外显子 20 插入。 |
|
|
| E1984 |
JBJ-09-063 hydrochloride
|
JBJ-09-063 hydrochloride 是一种突变选择性的 EGFR 变构抑制剂,对 EGFR L858R、EGFR L858R/T790M、EGFR L858R/T790M/C797S 和 EGFRLT/L747S 的 IC50 值分别为 0.147 nM、0.063 nM、0.083 nM 和 0.396 nM。JBJ-09-063 盐酸盐可显著抑制 EGFR、Akt 和 ERK1/2 的磷酸化。 |
|
|
| E0833 |
O-Demethyl-Gefitinib
|
O-Desmethyl gefitinib是gefitinib的主要活性代谢物,也是一种强效的EGFR (ErbB1/HER1)抑制剂,通过类似于gefitinib的机制抑制亚细胞的EGFR酪氨酸激酶,IC50分别为36和22 nM。 |
|
|
| E4680 |
MTX-531
|
MTX-531(NSC827271) 是同类首创的强效选择性 EGFR 和 PI3Kα 抑制剂,IC50 分别为 14.7 nM 和 6.4 nM。它可能在治疗头颈部鳞状细胞癌 (HSNCC)、鳞状肺癌和某些 EGFR/PI3K 驱动的三阴性乳腺癌方面发挥作用。 |
|
|
| S6809 |
SU5214
|
SU5214是 VEGF receptor 2 (VEGFR2/FLK-1) 和 EGFR 的抑制剂,对应的IC50值分别为14.8 µM 和 36.7 µM。 |
|
|
| E1844New |
STX-721
|
STX-721(Elticertinib)是一种不可逆的EGFR抑制剂,可抑制携带外显子20插入(ex20ins)突变的EGFR,选择性地抑制表达EGFR外显子20 A769_V770insASV和D770_N771insSVD的Ba/F3细胞的增殖,IC50分别为5.4和5.8 nM。 |
|
|
| S1025 |
Gefitinib (ZD1839)
|
Gefitinib是一种EGFR抑制剂,作用于NR6wtEGFR和NR6W细胞中的Tyr1173、Tyr992、Tyr1173和Tyr992,IC50 分别为37 nM、37nM、26 nM和57 nM。Gefitinib 可通过阻滞PI3K/AKT/mTOR信号通路来促进肺癌细胞的自噬和细胞凋亡。 |
- Nat Commun, 2025, 16(1):7287
- Nat Commun, 2025, 16(1):6451
- Cell Rep Med, 2025, 6(2):101929
|

|
| S7786 |
Erlotinib (CP-358774)
|
Erlotinib是一种EGFR抑制剂,IC50 为 2 nM,对EGFR的敏感性比对人c-Src 或 v-Ab高1000多倍。Erlotinib可诱导自噬。 |
- Nat Commun, 2025, 16(1):8932
- Cell Rep Med, 2025, S2666-3791(25)00272-1
- J Exp Clin Cancer Res, 2025, 44(1):290
|

|
| S1023 |
Erlotinib (CP-358774) Hydrochloride
|
Erlotinib HCl 是一种EGFR抑制剂,在无细胞试验中IC50为2 nM,对EGFR的选择性比对人c-Src或v-Abl高1000多倍。 |
- Nat Commun, 2026, 17(1)3228
- Cell Res, 2025, 10.1038/s41422-025-01110-x
- Nat Commun, 2025, 16(1):3591
|
|
| S2111 |
Lapatinib (GW572016)
|
Lapatinib 是一种有效的EGFR和ErbB2抑制剂,在无细胞试验中IC50分别为10.2和9.8 nM。Lapatinib 可诱导 ferroptosis 和细胞自噬。 |
- Cells, 2026, 15(4)383
- Nat Genet, 2025, 57(6):1452-1462
- Cancer Discov, 2025, 10.1158/2159-8290.CD-25-0605
|

|
| S7297 |
AZD9291 (Osimertinib)
|
Osimertinib (AZD9291, Mereletinib)是口服不可逆的,突变选择性EGFR抑制剂,在 LoVo细胞中对Exon 19 缺失的 EGFR,L858R/T790M EGFR,和 WT EGFR的IC50分别为12.92,11.44 和 493.8 nM。Phase 3。 |
- Acta Pharmacologica Sinica, November 15, 2017, 1512-1520
- Clinical Cancer Research, July 01, 2018, 3097-3107
- Cancer Biology & Medicine, December 09, 2024, 1156-1170
|

|
| S1011 |
Afatinib (BIBW2992)
|
Afatinib不可逆地抑制EGFR/ErbB,包括EGFR(wt),EGFR(L858R),EGFR(L858R/T790M),ErbB2(HER2)和ErbB4(HER4),在无细胞试验中IC50分别为0.5 nM,0.4 nM,10 nM,14 nM和1 nM。Afatinib可诱导自噬。 |
- Cell Res, 2025, 10.1038/s41422-025-01110-x
- Nat Genet, 2025, 10.1038/s41588-025-02158-6
- Cell Rep Med, 2025, 6(2):101929
|

|
| S1006 |
Saracatinib (AZD0530)
|
Saracatinib (AZD0530)是一种有效的Src抑制剂,无细胞试验中IC50为2.7 nM,对c-Yes, Fyn, Lyn, Blk, Fgr和Lck也具有活性;但对Abl和EGFR (L858R和L861Q)活性较低。Saracatinib可诱导自噬。Phase 2/3。 |
- Cancers (Basel), 2026, 18(7)1082
- J Clin Invest, 2025, e177599
- Cell Rep, 2025, 44(1):115109
|

|
| S5098 |
Gefitinib hydrochloride
|
Gefitinib (ZD1839) is an EGFR inhibitor with IC50s of 15.5 nM and 823.3 nM for WT EGFR and EGFR (858R/T790M), respectively. |
- Cell Death Dis, 2024, 15(8):555
- iScience, 2024, 27(10):110862
- Cancer Res, 2023, 83(2):316-331
|
|
| S7810 |
Afatinib Dimaleate
|
Afatinib Dimaleate不可逆地抑制 EGFR/HER2,包括 EGFR(wt),EGFR(L858R),EGFR(L858R/T790M) 和 HER2,IC50 分别为0.5 nM,0.4 nM,10 nM 和 14 nM;活性比对Gefitinib抵抗型L858R-T790M EGFR突变体高100倍。Afatinib (BIBW2992) Dimaleate 可诱导自噬。 |
- Cell, 2024, 187(3):712-732.e38
- Signal Transduct Target Ther, 2024, 9(1):153
- J Invest Dermatol, 2024, S0022-202X(23)03210-4
|

|
| S1028 |
Lapatinib Ditosylate
|
Lapatinib Ditosylate是一种有效的EGFR 和 ErbB2抑制剂,在无细胞试验中IC50分别为10.8和9.2 nM。 |
- Cell Death Dis, 2025, 16(1):118
- bioRxiv, 2025, 2025.05.29.656249
- NPJ Breast Cancer, 2024, 10(1):65
|

|
| S1143 |
AG-490
|
AG-490是一种EGFR抑制剂,在无细胞试验中IC50为0.1 μM,作用于EGFR比作用于ErbB2选择性高135倍,对JAK2也有抑制作用,对Lck,Lyn,Btk,Syk和Src没有抑制活性。 |
- Biomed Pharmacother, 2024, 180:117568
- Commun Biol, 2024, 7(1):1694
- NPJ Breast Cancer, 2024, 10(1):80
|

|
| S1046 |
Vandetanib (ZD6474)
|
Vandetanib是一种有效的 VEGFR2 抑制剂,在无细胞试验中IC50为40 nM。同时,也抑制VEGFR3和EGFR,IC50分别为110 nM 和500 nM。对PDGFRβ, Flt-1, Tie-2 和FGFR1作用效果不大,IC50为 1.1-3.6 μM, 对MEK, CDK2, c-Kit, erbB2, FAK, PDK1, Akt 和 IGF-1R几乎没有作用效果,IC50>10 μM。Vandetanib (ZD6474) 可增加凋亡并通过提高 reactive oxygen species (ROS) 的水平来诱导自噬。 |
- Frontiers in Pharmacology, May 10, 2024, 15:1345070
- Molecular Cancer Therapeutics, March 15, 2016, 503-11
- Oncotarget, June 14, 2016, 36101-36114
|

|
| S2150 |
Neratinib (HKI-272)
|
Neratinib是一种高度选择性的HER2和EGFR抑制剂,在无细胞试验中IC50分别为59 nM 和 92 nM;微弱抑制KDR和Src,对Akt,CDK1/2/4,IKK-2,MK-2,PDK1,c-Raf和c-Met没有显著的抑制作用。Phase 3。 |
- Nat Genet, 2025, 57(6):1452-1462
- Cancer Discov, 2025, 10.1158/2159-8290.CD-25-0605
- Neoplasia, 2025, 70:101246
|

|
| S1167 |
CP-724714
|
CP-724714是一种有效的,选择性HER2/ErbB2抑制剂,IC50为10 nM,无细胞试验中比作用于EGFR,InsR,IRG-1R,PDGFR,VEGFR2,Abl,Src,c-Met等选择性高640多倍。Phase 2。 |
- Front Immunol, 2024, 15:1335302
- Cells, 2024, 13(17)1452
- Cancer Res Commun, 2024, 10.1158/2767-9764.CRC-24-0221
|

|
| S5078 |
Osimertinib mesylate
|
Osimertinib mesylate (AZD9291) 是osimertinib的甲磺酸盐形式,是口服的第三代 epidermal growth factor receptor (EGFR) 酪氨酸激酶(TKI)抑制剂。 |
- Drug Resist Updat, 2025, 81:101237
- Oncogene, 2024, 10.1038/s41388-024-03220-z
- iScience, 2024, 27(10):110862
|
|
| S2728 |
AG-1478
|
AG-1478是一种选择性EGFR抑制剂,在无细胞试验中IC50为3 nM,对HER2-Neu,PDGFR,Trk,Bcr-Abl和InsR几乎没有作用活性。AG-1478(Tyrphostin AG-1478)通过靶向 phosphatidylinositol 4-kinase IIIα (PI4KA) 抑制脑心肌炎病毒encephalomyocarditis virus (EMCV)和丙型肝炎病毒hepatitis c virus (HCV)。 |
- PLoS Negl Trop Dis, 2026, 20(3):e0014153
- Cell Death Dis, 2025, 16(1):479
- J Stroke Cerebrovasc Dis, 2025, 34(6):108315
|

|
| S2727 |
Dacomitinib (PF-299804)
|
Dacomitinib是一种有效的,不可逆的泛ErbB抑制剂,最有效作用于EGFR,无细胞试验中IC50为6 nM。Dacomitinib 抑制 ERBB2 和 ERBB4 ,其对应的IC50值分别为45.7 nM和73.7 nM。Dacomitinib 可高效作用于携带EGFR或ERBB2突变型(耐Gefitinib)和携带EGFR T790M突变型的NSCLCs。Dacomitinib 可抑制细胞生长并诱导凋亡。Phase 2。 |
- Cell Rep Med, 2025, 6(8):102284
- Cell Oncol (Dordr), 2025, 10.1007/s13402-025-01097-y
- Nat Commun, 2024, 15(1):2742
|

|
| S1019 |
Canertinib (CI-1033)
|
Canertinib (CI-1033, PD183805)是一种泛ErbB抑制剂,作用于EGFR和ErbB2,IC50分别为1.5 nM和9.0 nM,但对PDGFR,FGFR,InsR,PKC,和CDK1/2/4等均无抑制活性。Phase 3。 |
- Journal of Cell Science, September 15, 2010, 3102-3111
- Cancer Research, March 1, 2021, 1398-1412
- Cells, January 26, 2022, 425
|

|
| S2192 |
Sapitinib (AZD8931)
|
Sapitinib (AZD8931)是一种可逆的,ATP竞争性EGFR,ErbB2和ErbB3抑制剂,无细胞试验中IC50分别为4 nM,3 nM和4 nM,作用于NSCLC细胞更有效,作用于ErbB家族比作用于MNK1和Flt选择性强100倍。Phase 2。 |
- Cell Death Dis, 2025, 16(1):457
- Biomolecules, 2025, 15(5)698
- Cell Death Dis, 2024, 15(12):912
|

|
| S8229 |
Brigatinib
|
Brigatinib是一种有效的、选择性的ALK抑制剂,IC50=0.6 nM;也是ROS1抑制剂,IC50=0.9 nM。它还能以相对较低的效力抑制IGF-1R、FLT3、FLT3(D835Y)和EGFR。 |
- Leukemia, 2025, 10.1038/s41375-025-02682-8
- Cell Death Dis, 2025, 16(1):194
- Toxicol Appl Pharmacol, 2025, 498:117310
|

|
| S2250 |
EGCG ((-)-Epigallocatechin Gallate)
|
EGCG ((-)-Epigallocatechin Gallate)是从绿茶中分离的一种有效的抗氧化剂Polyphenol flavonoid。抑制telomerase和DNA methyltransferase, 阻滞EGF受体和HER-2受体的激活,抑制脂肪酸合成酶以及谷氨酸脱氢酶的活性。 |
- Clin Mol Hepatol, 2025, 10.3350/cmh.2024.0694
- Cell Rep, 2025, 44(6):115799
- Front Pharmacol, 2024, 15:1403424
|

|
| S1342 |
Genistein
|
Genistein是一种来源于大豆的植物雌激素,是一种高特异性的蛋白酪氨酸激酶(PTK) 的抑制剂,在NIH-3T3细胞中通过EGF或者胰岛素介导而抑制有丝分裂,IC50分别为12μM和19 μM。 |
- J Immunother Cancer, 2025, 13(6)e011442
- Cell Rep, 2025, 44(6):115849
- International Journal of Polymeric Materials and Polymeric Biomaterials, 2025, 1563-1572
|
|
| S7284 |
Rociletinib (CO-1686)
|
Rociletinib (CO-1686, AVL-301)是一种不可逆的,选择性抑制突变型EGFR,在无细胞试验中作用于EGFRL858R/T790M和EGFRWT, Ki分别为21.5 nM和303.3 nM。Phase 2。 |
- iScience, 2024, 27(10):110862
- J Pers Med, 2022, 12(2)258
- Hum Cell, 2022, 10.1007/s13577-022-00671-y
|

|
| S7358 |
Poziotinib (NOV120101, HM781-36B)
|
Poziotinib是一种不可逆的泛-HER抑制剂,对HER1,HER2,和HER4的IC50分别为3.2 nM,5.3 nM和23.5 nM。Poziotinib可诱导凋亡和G1细胞周期阻滞。Phase 2。 |
- Autophagy, 2026, 1-27.
- Clin Cancer Res, 2025, 31(14):3002-3018
- Nat Biomed Eng, 2024, 10.1038/s41551-024-01273-9
|
|
| S1173 |
WZ4002
|
WZ4002是一种新型的,突变选择性EGFR抑制剂,作用于EGFR(L858R)/(T790M),在BaF3细胞系中IC50为2 nM/8 nM;对ERBB2磷酸化(T798I)没有抑制作用。 |
- J Extracell Vesicles, 2024, 13(7):e12494
- Cancer Metab, 2023, 11(1):20
- Cancer Gene Ther, 2022, 29(12):1878-1894
|

|
| S8362 |
Tucatinib (Irbinitinib, ONT-380, Arry-380)
|
Tucatinib (Irbinitinib, ONT-380, ARRY-380) 是一种具有口服活性、可逆的、ATP竞争性的的ErbB2(HER2)小分子抑制剂。在细胞实验中,ARRY-380对ErbB-2和p95 HER2的IC50s分别为8 nM和7 nM,对HER2的选择性是对EGFR的500倍。具有潜在的抗肿瘤活性。 |
- Biochem Pharmacol, 2026, 250(Pt 1):117962
- Cancer Cell, 2025, 43(4):776-796.e14
- Cancer Discov, 2025, 10.1158/2159-8290.CD-25-0605
|
|
| S1194 |
CUDC-101
|
CUDC-101是一种有效的,多靶点抑制剂,作用于HDAC,EGFR和HER2,IC50分别为4.4 nM, 2.4 nM,和15.7 nM,且抑制I/II型HDACs,但是对III型, Sir-type HDACs没有抑制作用。Phase 1。 |
- Oncotarget, 5 Jul 2017, 80109-80123
- ACS Medicinal Chemistry Letters, 25 Jul 2013, 858-862
- J Am Heart Assoc, 2025, 14(1):e037400
|

|
| S2205 |
DesMethyl Erlotinib (OSI-420) HCl
|
DesMethyl Erlotinib (OSI-420) HCl是Erlotinib的活性代谢产物(是一种EGFR抑制剂,IC50为2 nM)。 |
- Drug Metab Dispos, 2025, 53(5):100069
- J Pharm Biomed Anal, 2023, 236:115716
- Front Oncol, 2022, 12:939118
|

|
| S1079 |
PD153035 HCl
|
PD153035 HCl (SU-5271 HCl, AG1517 HCl, ZM 252868 HCl)是一种有效的,特异性EGFR抑制剂,无细胞试验中Ki和IC50分别为5.2 pM和29 pM;对PGDFR, FGFR, CSF-1, InsR和Src几乎无作用。 |
- Journal of Experimental & Clinical Cancer Research, July 29, 2020, 145
- Molecular Cancer Therapeutics, January 2015, 298-306
- Nature Communications, December 9, 2025, 1
|

|
| S2185 |
Allitinib tosylate
|
Allitinib (AST-1306, AST-6) 是一种新型,不可逆的EGFR和ErbB2抑制剂,IC50分别为0.5 nM和3 nM,对突变型EGFR T790M/L858R也有效,作用于过表达ErbB2的细胞更有效,作用于ErbB家族比作用于其他激酶选择性高3000倍。 |
- Mol Oncol, 2025, 10.1002/1878-0261.70096
- Hum Cell, 2024, 10.1007/s13577-024-01054-1
- Research Square, 2023, Version 1
|

|
| S1486 |
AEE788 (NVP-AEE788)
|
AEE788 (NVP-AEE788)是一种有效的EGFR和HER2/ErbB2抑制剂,IC50分别为2 nM和6 nM,对VEGFR2/KDR, c-Abl, c-Src,和Flt-1作用效果稍弱,对Ins-R, IGF-1R, PKCα和CDK1没有抑制作用。Phase 1/2。 |
- Cell Rep Med, 2022, 3(1):100492
- Genome Med, 2020, 18;12(1):17
- Oncotarget, 2019, 10(68):7185-7197
|

|
| S6546 |
PD153035
|
PD153035是一种特定的、有效的EGFR抑制剂,Ki值为5.2 pM。 |
- Biomed Pharmacother, 2023, 158:114176
- Cell Death Discov, 2023, 10.1038/s41420-023-01701-w
- Sci Signal, 2022, 15(728):eabj6915
|
|
| S1056 |
AC480 (BMS-599626)
|
AC480 (BMS-599626)是一种选择性的,高效效的HER1和HER2抑制剂,IC50分别为20 nM和30 nM。比对HER4效果强8倍左右,而比对VEGFR2, c-Kit, Lck, MET等的作用强100倍以上。Phase 1。 |
- Communications Biology, 2025, Article number: 1350
- Int J Mol Sci, 2022, 23(22)14023
- Genome Med, 2020, 18;12(1):17
|

|
| S1392 |
Pelitinib (EKB-569)
|
Pelitinib (EKB-569) 是一种有效的,不可逆 EGFR 抑制剂,IC50为38.5 nM。Pelitinib (EKB-569) 还轻微抑制 Src、MEK/ERK 和 ErbB2 ,对应的IC50值分别为282 nM、800 nM和1255 nM。Phase2。 |
- Cancer Cell, 2022, S1535-6108(22)00312-9
- Cancer Discov, 2022, 12(5):1378-1395
- Cell Oncol (Dordr), 2022, 45(4):601-619
|

|
| S2922 |
Icotinib (BPI-2009H)
|
Icotinib (BPI-2009H)是一种有效的,特异性EGFR抑制剂,IC50为5 nM,包括EGFR, EGFR(L858R), EGFR(L861Q), EGFR(T790M)和EGFR(T790M, L858R)。Phase 4。 |
- iScience, 2024, 27(10):110862
- Am J Cancer Res, 2024, 14(1):33-51
- Cell Rep Med, 2022, 3(1):100492
|

|
| S2867 |
WHI-P154
|
WHI-P154是一种有效的JAK3抑制剂,IC50为1.8 μM,对JAK1和JAK2没有抑制活性,也抑制EGFR, Src, Abl, VEGFR和MAPK,抑制Stat3而非Stat5磷酸化。 |
- Nat Commun, 2023, 14(1):6908
- Int J Mol Sci, 2023, 10.3390/ijms241813863
- Int J Mol Sci, 2023, 24(18)13863
|

|
| E0822 |
AST-1306
|
AST-1306 (Allitinib)是一种口服活性和不可逆的EGFR、ErbB2和ErbB4抑制剂,IC50s分别为0.5,3和0.8 nM。 |
- Mol Oncol, 2025, 10.1002/1878-0261.70096
- Molecular Oncology, 2025, 3205-3222
- Human Cell, 2024, 1170-1183
|
|
| S8724 |
Lazertinib (YH25448)
|
Lazertinib (YH25448,GNS-1480)是一种有效的、对突变型具有高选择性的、不可逆的EGFR抑制剂,对Del19/T790M、L858R/T790M、Del19、L85R和野生型EGFR的IC50分别为1.7 nM, 2 nM, 5 nM, 20.6 nM和76 nM。对ErbB2和ErbB4的IC50值更高。 |
- Cell Rep Med, 2025, 6(2):101929
- Cells, 2025, 14(17)1386
- Front Oncol, 2025, 15:1533059
|
|
| S2755 |
Varlitinib
|
Varlitinib (ARRY334543) 是一种有效的,选择性ErbB1(EGFR)和ErbB2(HER2)抑制剂,IC50分别为7 nM和2 nM。Phase 2。 |
- J Transl Med, 2023, 21(1):89
- Cell Rep Med, 2022, 3(1):100492
- Pharmaceutics, 2019, 11(11)
|

|
| S0711 |
Canertinib dihydrochloride
|
Canertinib (CI-1033, PD-183805, compound 18) dihydrochloride 是一种有效且不可逆的 epidermal growth factor receptor (EGFR) tyrosine kinase 的抑制剂。Canertinib dihydrochloride 可抑制细胞 EGFR 和 ErbB2 自磷酸化,IC50值分别为 7.4 nM和 9 nM。 |
- Front Microbiol, 2022, 13:968872
- Acta Physiol (Oxf), 2021, 10.1111/apha.13661
- Sci Rep, 2017,
|
|
| S7926 |
Lifirafenib (BGB-283)
|
Lifirafenib (BGB-283, Beigene-283) 有效地抑制RAF激酶家族和EGFR活性,在生化实验检测中,其对重组BRAF(V600E)激酶区域、EGFR和EGFR(T790M/L858R)突变体的IC50分别为23、29、495 nM。 |
- Cancer Research Communications, 2025, nan
- Sci Adv, 2023, 9(35):eade7486
- Mol Cancer Ther, 2022, MCT-22-0004
|
|
| S7971 |
Zorifertinib (AZD3759)
|
Zorifertinib (AZD3759)是一种有效的,具有口服活性的,CNS-渗透性EGFR抑制剂,对EGFR (WT),EGFR (L858R),和 EGFR (exon 19Del)的IC50分别为0.3 nM,0.2 nM,和 0.2 nM。Phase 1。
|
- Frontiers in Immunology, June 6, 2025, 1606125
- Drug Metabolism and Disposition, April 2019, 393-404
- Drug Des Devel Ther, 2024, 18:1175-1188
|
|
| S7039 |
PD168393
|
PD168393是一种不可逆的EGFR抑制剂,IC50为0.70 nM,不可逆烷基化Cys-773;抑制insulin, PDGFR, FGFR和PKC活性。 |
- Elife, 2024, 13e85914
- FEBS Open Bio, 2023, 10.1002/2211-5463.13653
- J Neurosci Res, 2021, 10.1002/jnr.24909
|

|
| S7298 |
AZ5104
|
AZ5104,是AZD-9291的脱甲基代谢物,是一种有效的EGFR抑制剂,作用于EGFR (L858R/T790M),EGFR (L858R),EGFR (L861Q),和EGFR (野生型),IC50分别为<1 nM,6 nM,1 nM,和25 nM。Phase 1。 |
- Genes & Diseases, 2023, 2241-2244
- J Pharm Biomed Anal, 2020, 177:112871
- Clin Cancer Res, 2019, 25(8):2575-2587
|
|
| S8294 |
Olmutinib (BI 1482694)
|
Olmutinib (BI 1482694) 是一种新型的EGFR突变特效性酪氨酸激酶抑制剂、布鲁顿氏酪氨酸激酶抑制剂。 |
- iScience, 2024, 27(10):110862
- Br J Pharmacol, 2022, 179(11):2754-2770
- Ther Adv Med Oncol, 2022, 14:17588359221079125
|
|
| S7824 |
Nazartinib (EGF816)
|
Nazartinib (EGF816, NVS-816)是共价的、不可逆的、对突变型EGFR具有选择性的抑制剂,对突变型EGFR(L858R, ex19del)和T790M具有纳摩尔级别的抑制作用,相对于野生型EGFR,其对突变型EGFR更具特异性。 |
- Translational Oncology, March 2019, 705-713
- Cancer Cell, July 11, 2022, 754-767.e6
- Cancer Cell, 2022, 40(7):754-767.e6
|
|
| S8852 |
Pyrotinib dimaleate
|
Pyrotinib dimaleate是一种有效的、具有选择性的、不可逆的 EGFR 和 HER2 双重酪氨酸激酶抑制剂,其对应的IC50值分别为0.013 μM和0.038 μM。 |
- Cancer Research, 2026, nan
- Cancer Research, 2025 Oct 15, 3983-3998
- Cancer Research, 2025, 3983-3998
|
|
| S7000 |
AP26113-analog (ALK-IN-1)
|
AP26113-analog (ALK-IN-1) 是AP26113的类似物,是有效的、选择性的ALK抑制剂,同时也是EGFR的抑制剂。 |
- ChemMedChem, 2017, 1219-1230
- Clin Cancer Res, 2015, 21(1):166-74
- Clin Cancer Res, 2014, 20(22):5686-96
|

|
| S2784 |
TAK-285
|
TAK-285是一种新型,HER2和EGFR(HER1)双重抑制剂,IC50分别为17 nM和23 nM,作用于HER1/2比作用于HER4选择性高10倍以上,对MEK1/5, c-Met, Aurora B, Lck, CSK等作用效果稍弱。Phase 1。 |
- Oncotarget, 2020, 11(46):4224-4242
- Oncotarget, 2020, 4224-4242
- Mol Pharmacol, 2019, 95(5):528-536
|
|
| S7557 |
CL-387785 (EKI-785)
|
CL-387785 (EKI-785, WAY-EKI 785)是一种不可逆的选择性EGFR抑制剂,IC50为370 pM。
|
- iScience, 2025, 28(2):111708
- Stem Cell Reports, 2023, 10.1016/j.stemcr.2023.03.011
- Nature, 2017, 550(7675):270-274
|

|
| S6813 |
Mobocertinib (TAK788)
|
Mobocertinib (TAK788, AP32788) 是一种研究用的TKI,是有效的、选择性的 epidermal growth factor receptor (EGFR) 和 HER2 突变体的临床前抑制剂。Mobocertinib (TAK788) 是一种抗肿瘤药。 |
- Int J Mol Sci, 2024, 25(7)3992
- J Thorac Oncol, 2023, S1556-0864(23)00797-9
- Genes & Diseases, 2023, 2241-2244
|
|
| S3759 |
Norcantharidin
|
Norcantharidin (Endothall anhydride) is a synthetic anticancer compound which is a dual inhibitor for c-Met and EGFR in human colon cancers. |
- Frontiers in Pharmacology, November 3, 2022, 1019478
- Frontiers in Pharmacology, November 02, 2022, 1019478
- Molecular Medicine Reports, March 8, 2024, 71
|
|
| S8814 |
zipalertinib (TAS6417)
|
zipalertinib (TAS6417)是一种新型的EGFR抑制剂,靶向EGFR突变体,对野生型EGFR抑制效力较低,IC50范围为1.1 ± 0.1到8.0 ± 1.1 nmol/L。 |
- iScience, 2024, 108839
- iScience, 2024, 27(2):108839
- STAR Protocols, 2024, 102987
|
|
| S2554 |
Daphnetin
|
Daphnetin是天然香豆素衍生物,是一种蛋白激酶抑制剂,抑制EGFR, PKA和PKC,IC50分别为7.67 μM, 9.33 μM和25.01 μM,且具有抗炎和抗氧化剂活动性。 |
- Sci Rep, 2021, 11(1):11491
- Acta Neuropathol, 2020, 10.1007/s00401-020-02226-7
- Int Immunopharmacol, 2019, 72:195-203
|

|
| S0360 |
EGFR Inhibitor
|
EGFR inhibitor 是一种可透过细胞的4,6-二取代嘧啶化合物,是一种具有高度选择性的 EGFR kinase 的抑制剂,其IC50值为21 nM。EGFR inhibitor 可直接使 microtubules 解聚,可作为研究EGFR途径和微管动力学的化学探针。 |
- Cell Rep, 2022, 40-8:111223
- Frontiers in Pharmacology, 2022, 804327
- Cancers (Basel), 2022, 14(19)4910
|
|
| S8817 |
Almonertinib (HS-10296)
|
Almonertinib (HS-10296)是一种EGFR激活突变和对EGFR T790M具有耐受性突变的抑制剂,其对野生型EGFR仅具有有限活性。 |
- Cells, 2025, 14(17)1386
- iScience, 2024, 27(10):110862
- Biomedicines, 2024, 12(7)1412
|
|
| S1179 |
WZ8040
|
WZ8040是一种新型的,突变选择性的,不可逆的EGFRT790M抑制剂,对ERBB2磷酸化(T798I)没有抑制作用。 |
- J Biol Chem, 2025, 301(8):110454
- Biochemistry, 2018, 57(8):1369-1379
- Nat Commun, 2016, 7:10690
|

|
| E4819 |
(E/Z)-Afatinib
|
(E/Z)-Afatinib (Gilotrif, BIBW2992, Boehringer Ingelheim)是第二代酪氨酸激酶抑制剂(TKI),对EGFR和HER2具有不可逆抑制作用,对野生型EGFR、EGFR-L858R、HER2、ErbB4的IC50分别为0.5 nM、0.4 nM、14 nM和1 nM。 |
- Cell Death & Disease, 2021, 728
- Gynecologic Oncology, 2019 Apr, 158-164
- Bioscience Reports, 2018, BSR20181964
|
|
| S6868 |
Furmonertinib Mesylate (AST2818, Alflutinib)
|
Alflutinib (Furmonertinib) mesylate是第三代 epidermal growth factor receptor (EGFR) 的抑制剂,可抑制EGFR敏感突变和T790M突变。Alflutinib (AST2818) 主要由CYP3A4代谢,也是有效的 CYP3A4 的诱导剂,EC50值为0.25 μM。 |
- Cells, 2025, 14(17)1386
- Biomedicines, 2024, 12(7)1412
- Int J Mol Sci, 2023, 24(18)13972
|
|
| S2406 |
Chrysophanic Acid
|
Chrysophanic Acid (Chrysophanol) 是Dianella longifolia中的一种天然蒽醌,是一种EGFR/mTOR通道抑制剂。 |
- Cell Proliferation, June 29, 2020, e12871
- International Journal of Oncology, February 11, 2025, 20
- Int J Oncol, 2025, 66(3)20
|

|
| S9786 |
Tuxobertinib (BDTX-189)
|
Tuxobertinib (BDTX-189)是一种有效的 EGFR 和 HER2 致癌突变的变构性抑制剂,对EGFR、HER2、BLK 和 RIPK2的Kd值分别为0.2 nM、0.76 nM、13 nM和1.2 nM。BDTX-189 具有抗癌活性。 |
- Molecular Pharmacology, 2024 Jan 10, 97-103
- Chem Biol Interact, 2024, 395:111033
- J Transl Med, 2023, 21(1):89
|
|
| S4667 |
Lidocaine hydrochloride
|
Lidocaine hydrochloride (Lidothesin, Lignocaine, Xyloneural)是一种 epidermal growth factor receptor (EGFR) 的抑制剂,对人舌癌细胞具有抗增殖作用。Lidocaine hydrochloride 是一种局部麻醉剂及心脏抑制剂,被用作抗心律失常剂。 |
- Cancer Research, July 1, 2021, 3580-3592
- J Immunother Cancer, 2024, 12(11)e009805
- Exp Ther Med, 2021, 21(5):424
|
|
| S8036 |
Butein
|
Butein,一种从漆树中分离的植物多酚,可以抑制酪氨酸激酶,NF-κB,STAT3和EGFR活化。 |
- Scientific Reports, 2022, 20644
- American Journal of Cancer Research, 2020, 3721-3736
- Am J Cancer Res, 2020, 10(11):3721-3736
|
|
| S8242 |
EAI045
|
EAI045是一种靶向特定耐药性的EGFR突变体的变构抑制剂。对野生型受体无作用。 |
- bioRxiv, August 22, 2021, nan
- EJNMMI Res, 2024, 14(1):19
- Pharmazie, 2018, 73(11):630-634
|
|
| E4886 |
HS-10296 hydrochloride
|
HS-10296 hydrochloride(Almonertinib (hydrochloride), HS-10296 HCl) 是一种不可逆的第三代 EGFR 酪氨酸激酶 抑制剂,对 EGFR 敏感突变、T790M 抗性突变具有高选择性。它对 T790M、T790M/L858R 和 T790M/Del19 具有抑制活性,IC50 分别为 0.37 nM、0.29 nM 和 0.21 nM。它还具有抗肿瘤活性。 |
- Cells, 2025, 1386
- Cancer Medicine, 2025, e70827
- Advanced Science, 2024, e2405130
|
|
| S8741 |
Avitinib (Abivertinib)
|
Avitinib (Abivertinib)是不可逆的EGFR抑制剂,对于突变型EGFR具有选择性,对EGFR L858R/T790M双突变体的IC50为0.18 nM,是对野生型EGFR效力的近43倍(IC50=7.68 nM for WT EGFR)。Avitinib具有抗肿瘤活性和耐受性毒性。 |
- iScience, 2024, 27(10):110862
- Microsyst Nanoeng, 2023, 9:57
- , 2022, 10.21203/rs.3.rs-2146794/v1
|
|
| S1170 |
WZ3146
|
WZ3146是一种突变选择性的,不可逆EGFR(L858R)和EGFR(E746_A750)抑制剂,IC50分别为2 nM和2 nM;对ERBB2磷酸化(T798I)没有抑制作用。 |
- Sci Rep, 2024, 14(1):22895
- Aging (Albany NY), 2020, 12(19):19022-19044
|

|
| S2895 |
Tyrphostin 9
|
Tyrphostin 9 (SF 6847, RG-50872) 是第一个EGFR抑制剂,IC50为460 μM,作用于PDGFR更有效,IC50为0.5μM。 |
- PLoS One, June 2, 2020, e0233993
- Cell Rep, 2022, 38(10):110475
|
|
| S7206 |
CNX-2006
|
CNX-2006是一种新型,不可逆,突变选择性的EGFR抑制剂,IC50为<20 nM,对野生型EGFR有微弱的抑制作用。 |
- Nat Nanotechnol, 2021, 10.1038/s41565-021-00872-w
- Cancer Science, 2016, 461-468
- Cancer Sci, 2016, 107(4):461-8
|

|
| E5906 |
Sunvozertinib
|
Sunvozertinib (DZD9008) 是一种口服、强效且选择性的 ErbBs(EGFR 或 Her2) 抑制剂,特别是 ErbBs 突变形式。它还可抑制 EGFR 外显子 20 NPH 插入、EGFR 外显子 20 ASV 插入、EGFR L858R 和 T790M 突变、Her2 外显子 20 YVMA 和 EGFR WT A431,IC50 值分别为 20.4 nM、20.4 nM、1.1 nM、7.5 nM 和 80.4 nM。 |
- Journal of Thoracic Oncology, 2024, 71-79
- bioRxiv , 2023, 10.1101/2023.07.27.550902
|
|
| S6541 |
MTX-211
|
MTX-211是PI3K和EGFR的双重抑制剂。 |
- Int J Mol Sci, 2024, 25(10)5160
- Int J Mol Sci, 2023, 24(8)7608
|
|
| S8412 |
Naquotinib(ASP8273)
|
Naquotinib (ASP8273)是一种具有口服活性的、不可逆的、对突变体具有选择性的epidermal growth factor receptor (EGFR)抑制剂,具有潜在的抗肿瘤活性。 |
- ACS Infectious Diseases, 2019, 1764-1771
- Expert Opin Emerg Drugs, 2018, 10.1080/14728214.2018.1558203
Share on FacebookShare on TwitterShare on Google+
|
|
| S2752 |
HER2-Inhibitor-1
|
HER2-Inhibitor-1是ARRY-380的类似物。ARRY-380是一种有效的,选择性的HER2抑制剂,IC50为8 nM,等效作用于截短的p95-HER2,作用于HER2比作用于EGFR选择性高500倍。 |
- Communications Biology, 2021, 762
- Commun Biol, 2021, 4(1):762
|

|
| S8009 |
AG-18
|
AG-18 (RG-50810, Tyrphostin A23, TX 825)抑制EGFR,IC50为35 μM。 |
- Reproduction, 2016, 151(2):179-85
|
|
| S6899 |
Licochalcone D
|
Licochalcone D (Lico D, LCD, LD) 是一种从中草药甘草 Glycyrrhiza inflata 中分离出的类黄酮,具有抗氧化、抗炎和抗癌的特性。Licochalcone D 可抑制LPS信号通路中NF-κB p65的磷酸化。Licochalcone D 可抑制 JAK2、EGFR 和 Met (c-Met) 活性,并诱导ROS依赖性的凋亡。Licochalcone D 还可诱导 caspases 的活化和 poly (ADP-ribose) polymerase (PARP) 的裂解。 |
- Frontiers in Pharmacology, September 18, 2025, 16:1573882
|
|
| S6525 |
AG 555
|
AG-555 (Tyrphostin B46)是酪氨酸激酶抑制剂,可与拓扑异构酶I直接相互作用,因而阻止DNA松弛。它抑制EGFR,IC50为0.7 μM。 |
- Tesi Doctoral, Universitat Autònoma de Barcelona, 2017, 99
|
|
| E1176 |
BLU-945
|
BLU-945是一种强效、高选择性、可逆、口服活性表皮生长因子受体酪氨酸激酶(EGFR)抑制剂(TKIs)。BLU-945可用于肺癌包括非小细胞肺癌(NSCLC)的研究。 |
- Eur J Pharm Sci, 2025, S0928-0987(25)00335-5
|
|
| S9711 |
CH7233163
|
CH7233163 是一种 EGFR-tyrosine kinase 的非共价ATP竞争性抑制剂,对有EGFR-Del19/T790M/C797S的肿瘤具有抗肿瘤活性。 |
- bioRxiv , 2023, 10.1101/2023.07.27.550902
|
|
| E1791 |
Zongertinib (BI 1810631)
|
Zongertinib (BI 1810631, BI 764532)是一种有效的选择性人HER2和EGFR酪氨酸激酶抑制剂,共价结合野生型和突变的HER2受体,包括外显子20插入突变,同时保留EGFR信号;口服Zongertinib治疗HER2异常阳性的实体瘤有效且耐受。 |
- Cancer Discov, 2025, 10.1158/2159-8290.CD-25-0605
|
|
| S6698 |
TQB3804
|
TQB3804是一种选择性有效的 EGFR 激酶抑制剂,对于 EGFRd746-750/T790M/C797S、EGFRL858R/T790M/C797S、EGFRd746-750/T790M、EGFRL858R/T790M 和 EGFRWT的IC50值分别为0.46 nM、0.13 nM、0.26 nM、0.19 nM 和 1.07 nM。TQB3804 (EGFR-IN-7) 也具有抗肿瘤活性。 |
- Signal Transduction and Targeted Therapy, 2025, 389
- Signal Transduct Target Ther, 2025, 10(1):389
|
|
| S8921 |
BI-4020
|
BI-4020 是一种口服活性的 EGFR 的非共价抑制剂,其对 EGFRdel19 T790M C797S 的在BaF3细胞中的生物标志物效力和抗增殖效力对应的IC50分别为0.6 nM和0.2 nM。 |
- Biomedicines, 2024, 12(7)1412
- Biomedicines, 2024, 1412
|
|
| E0368 |
(S)-Sunvozertinib ((S)-DZD9008)
|
(S)-Sunvozertinib ((S)-DZD9008) 是一种口服、有效、不可逆、野生型选择性 EGFR 抑制剂,可抑制 EGFR 或 HER2 Exon20ins 和其他突变体,对突变型 EGFR 的 IC50 范围为 0.4 nM 至 2.1 nM。 |
- J Thorac Oncol, 2023, S1556-0864(23)00797-9
|
|
| E1919 |
Befotertinib mesylate
|
Befotertinib mesylate (D-0316 mesylate) 是第三代EGFR酪氨酸激酶抑制剂(TKI),可用于EGFR T790M阳性非小细胞肺癌(NSCLC)的研究。 |
- Cells, 2025, 14(17)1386
|
|
| E2399 |
Falnidamol
|
Falnidamol (BIBX1382)是一种选择性的表皮生长因子受体(epidermal growth factor receptor, EGFR)抑制剂,通过抑制丝裂原活化蛋白激酶(mitogen-activated protein kinases, MAPKs)信号传导,在体外和体内对人头颈部鳞状细胞癌发挥抗癌作用。 |
- Free Radical Biology and Medicine, 2022, 106-124
|
|
| E4675 |
Rezivertinib
|
Rezivertinib(BPI-7711) 是一种有效的第三代 EGFR 酪氨酸激酶 抑制剂,可同时靶向 EGFR 敏感突变和 EGFR T790M 突变。此外,它具有出色的中枢神经系统渗透性,并具有抗肿瘤活性。 |
- Cells, 2025, 14(17)1386
- Cells, 2025, 1386
|
|
| S9414 |
Cyasterone
|
Cyasterone is the main phytoecdysteroid component found in Cyathula capitata. It is a natural EGFR inhibitor and maybe a promising anti-cancer agent. |
- Chin Med, 2023, 18(1):136
|
|
| S6897 |
Epertinib hydrochloride
|
Epertinib hydrochloride (S-222611 hydrochloride) 是有一种效的、口服活性的、可逆的、选择性的 tyrosine kinase 抑制剂,针对 EGFR、HER2 和 HER4,对应的IC50值分别为1.48 nM、7.15 nM和2.49 nM。Epertinib hydrochloride (S-222611 hydrochloride) 具有抗肿瘤活性。 |
- Biomedicine & Pharmacotherapy, 2024, 117542
|
|
| E4904 |
Dacomitinib hydrate
|
Dacomitinib hydrate(PF-00299804, PF-804)是一种口服给药、高度选择性的不可逆抑制剂,靶向作用于泛ERBB受体,对EGFR的IC50为6.0 nM,对ERBB2的IC50为45.7 nM,对ERBB4的IC50为73.7 nM。此外,它还表现出抗肿瘤活性。 |
- McGill University, Montreal, 2021,
|
|
| E2980 |
4-AMino-1-phenylpyrazolo[3,4-d]pyriMidine
|
4-AMino-1-phenylpyrazole[3,4-d]pyriMidine(PP 3)是一种 EGFR 酪氨酸激酶抑制剂,IC50 为 2.7 μM。 它是 Src 激酶抑制剂 PP 2 的阴性对照。 |
- Molecular Biology of the Cell, March 01, 2021, 376-390
- Molecular Biology of the Cell, 2021, 376-390
|
|
| E4449 |
AG 825
|
AG-825 (Tyrphostin AG-825) 是一种有效的选择性 ErbB2 抑制剂,IC50 值为 0.15 µM。 它还抑制 PDGFR 自磷酸化,IC50 值为 40 µM。 它具有抗癌活性并显着诱导癌细胞凋亡。 |
|
|
| E1749 |
limertinib
|
Limertinib (ASK120067) 是一种不可逆的第三代 EGFRT790M 抑制剂,IC50 为 0.3 nM,选择性优于 EGFRWT< /b> IC50 为 6.0 nM。 它在非小细胞肺癌 (NSCLC) 肿瘤细胞中表现出有效的抗增殖活性。 |
|
|
| S6805 |
Tyrphostin AG-528
|
Tyrphostin AG-528 (Tyrphostin B66) 是一种 epidermal growth factor receptors (EGFR) 和 ErbB2/HER2 的有效抑制剂,其IC50值分别为4.9 μM和2.1 μM。Tyrphostin AG-528 具有抗癌活性。 |
|
|
| E5912 |
Asandeutertinib
|
Asandeutertinib (Osimertinib-d3, AZD-9291-d3) 是一种口服、ATP 竞争性、不可逆、氘化的第三代表皮生长因子受体酪氨酸激酶抑制剂 (EGFR TKI),也是osimertinib 的氘稳定化衍生物,具有治疗晚期 EGFR 突变 NSCLC 的潜力。 |
|
|
| S6509 |
AG 494
|
AG-494是EGFR受体自身磷酸化的抑制剂,IC50为1.2 μM。它抑制依赖于EGF的细胞生长,IC50为6 μM。 |
|
|
| S0736 |
AG 556
|
AG-556是一种有效的EGFR酪氨酸抑制剂,IC50为5µM,可抑制EGF诱导的HER14细胞生长。 |
|
|
| S6523 |
RG14620
|
RG14620 (Tyrphostin RG14620)是一种EGFR抑制剂,可直接抑制ABCG2/BCRP的传送功能。 |
|
|
| E1877 |
Pebezertinib (BLU-451, LNG-451)
|
Pebezertinib 是一种中枢神经系统(CNS)渗透性,保留野生型EGFR功能,针对EGFR exon 20 insertion (Ex20ins)的共价抑制剂,IC50为7-78 nM,具有强大的抗肿瘤功效。 |
|
|
| S0070 |
Gefitinib-based PROTAC 3
|
Gefitinib-based PROTAC 3 通过连接器将EGFR结合元件与VHL配体偶联,可诱导 EGFR 及其突变体的降解,在HCC827(第19外显子)和H3255(L858R)中的DC50值分别为11.7 nM和22.3 nM。 |
|
|
| S0324 |
Tyrphostin AG30 (AG30)
|
Tyrphostin AG30 (AG30)是一种有效的选择性EGFR酪氨酸激酶抑制剂,选择性抑制c-ErbB诱导的自我更新,并能够抑制c-ErbB在原代成红细胞中对STAT5的激活。 |
|
|
| E0344 |
ErbB2 inhibitor
|
ErbB-2 Inhibitor是一种EGFR、ErbB2和ErbB4抑制剂,IC50分别为0.017 μM, 0.08 μM, 1.91 μM。 |
|
|
| E4597 |
Avitinib maleate
|
Avitinib maleate(Abivertinib maleate, AC0010 maleate, AC0010MA) 是第三代、不可逆且口服有效的 EGFR 选择性抑制剂。其 IC50 值分别为 0.18 nM、0.18 nM、7.68 nM,针对 EGFR L858R、EGFR T790M 和野生型 EGFR。Avitinib maleate还可作为 BTK 抑制剂,诱导套细胞淋巴瘤中的 BTK 凋亡。 |
|
|
| E4664 |
Osimertinib dimesylate
|
Osimertinib dimesylate (Mereletinib dimesylate, AZD-9291 dimesylate) 是一种强效、突变选择性、不可逆的 EGFR 激酶活性抑制剂,对 EGFRL858R 和 EGFRL858R/T790M 的 IC50 分别为 12 nM和 1 nM。它在异种移植和转基因 T790M EGFR 突变模型中表现出抗肿瘤活性,支持其在非小细胞肺癌 (NSCLC) 研究中的应用。 |
|
|
| S2733 |
AV-412 free base
|
AV-412 free base (MP-412 free base)是一种EGFR抑制剂,对EGFR,EGFRL858R,EGFRT790M,EGFRL858R/T790M和ErbB2的IC50分别为0.75,0.51,0.79,2.30和19 nM。 |
|
|
| S8920 |
(Rac)-JBJ-04-125-02
|
(Rac)-JBJ-04-125-02 (JBJ-04-125-02 racemate) 是一种 Epidermal growth factor receptor (EGFR) 的突变体选择性的变构抑制剂,具有潜在的抗癌活性。 |
|
|
| S6881 |
JND3229
|
JND3229 是一种有效且可逆的EGFRC797S抑制剂,IC50为5.8 nM。JND3229还有效抑制EGFRL858R/T790M和EGFRWT,IC50值分别为30.5 nM 和6.8 nM。 |
|
|
| S1054 |
AG99
|
AG99 (Tyrphostin 46,Tyrphostin A46,Tyrphostin B40)是一种有效、选择性的EGFR抑制剂。 |
|
|
| S8584 |
Theliatinib (HMPL-309)
|
Theliatinib (HMPL-309)是一种高效的EGFR抑制剂,对EGFR的Ki值为0.05 nM,对EGFR和EGFR T790M/L858R突变体的IC50值分别为3 nM和22 nM。它对EGFR的选择性是对其他72种激酶的50倍以上。 |
|
|
| E6000 |
BIEGi-1
|
BIEGi-1 是一种强效且选择性的EGFR二元抑制剂,可有效阻断 EGFR 催化的核苷酸交换并破坏 EGFR-Rheb 相互作用。它能够强效抑制 EGFR 突变癌细胞中的 EGFR 激酶活性和 mTORC1 信号传导。BIEGi-1 表现出显著的抗增殖作用,对 PC9 的 IC50 为 17 nM,对 HCC827 的 IC50 为 20 nM。 |
|
|
| S0151 |
AG-1557
|
AG-1557 是一种特异性的 epidermal growth factor receptor (EGFR) 酪氨酸激酶的ATP竞争性抑制剂。 |
|
|
| S2115 |
RG 13022
|
RG 13022 (Tyrphostin RG13022)抑制免疫沉淀反应中EGF receptor的自磷酸化反应,IC50为4 µM。 |
|
|
| E4884 |
Icotinib Hydrochloride
|
Icotinib hydrochloride (BPI-2009H) 是一种高效、选择性 表皮生长因子受体酪氨酸激酶抑制剂 (EGFR-TKI),IC50 为 5 nM,用于治疗非小细胞肺癌 (NSCLC)。 |
|
|
| E6528New |
BBT-176
|
BBT-176 是一种首创的可逆性第四代EGFR TKI,对奥希替尼耐药突变具有强效活性,在 Ba/F3 细胞中 IC50 分别为 49 nM (Del19/T790M/C797S)、202 nM (L858R/T790M/C797S)、42 nM (Del19/C797S) 和 183 nM (L858R/C797S)。 |
|
|
| E1478 |
BDTX-1535
|
BDTX-1535 (EGFR-IN-76) 是一种口服、高效、选择性和不可逆的 EGFR 抑制剂,可以穿透血脑屏障,具有治疗胶质母细胞瘤或非小细胞瘤的潜力 细胞肺癌。 |
|
|
| E4997 |
Sevabertinib (BAY 2927088)
|
BAY 2927088(Sevabertinib)是首个非共价、强效、口服、选择性、可逆性 酪氨酸激酶 抑制剂,在临床前模型中可强效抑制突变型 HER2 和突变型 EGFR。该药物靶向非小细胞肺癌 (NSCLC) 患者的外显子 20 插入。 |
|
|
| E1984 |
JBJ-09-063 hydrochloride
|
JBJ-09-063 hydrochloride 是一种突变选择性的 EGFR 变构抑制剂,对 EGFR L858R、EGFR L858R/T790M、EGFR L858R/T790M/C797S 和 EGFRLT/L747S 的 IC50 值分别为 0.147 nM、0.063 nM、0.083 nM 和 0.396 nM。JBJ-09-063 盐酸盐可显著抑制 EGFR、Akt 和 ERK1/2 的磷酸化。 |
|
|
| E0833 |
O-Demethyl-Gefitinib
|
O-Desmethyl gefitinib是gefitinib的主要活性代谢物,也是一种强效的EGFR (ErbB1/HER1)抑制剂,通过类似于gefitinib的机制抑制亚细胞的EGFR酪氨酸激酶,IC50分别为36和22 nM。 |
|
|
| E4680 |
MTX-531
|
MTX-531(NSC827271) 是同类首创的强效选择性 EGFR 和 PI3Kα 抑制剂,IC50 分别为 14.7 nM 和 6.4 nM。它可能在治疗头颈部鳞状细胞癌 (HSNCC)、鳞状肺癌和某些 EGFR/PI3K 驱动的三阴性乳腺癌方面发挥作用。 |
|
|
| S6809 |
SU5214
|
SU5214是 VEGF receptor 2 (VEGFR2/FLK-1) 和 EGFR 的抑制剂,对应的IC50值分别为14.8 µM 和 36.7 µM。 |
|
|
| E1844New |
STX-721
|
STX-721(Elticertinib)是一种不可逆的EGFR抑制剂,可抑制携带外显子20插入(ex20ins)突变的EGFR,选择性地抑制表达EGFR外显子20 A769_V770insASV和D770_N771insSVD的Ba/F3细胞的增殖,IC50分别为5.4和5.8 nM。 |
|
|







































