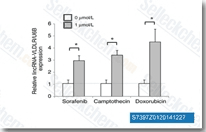
| S7397 |
Sorafenib (BAY 43-9006)
|
Sorafenib是Raf-1和B-Raf的多重激酶抑制剂,无细胞试验中IC50分别为6 nM和22 nM。Sorafenib 还可抑制VEGFR-2、VEGFR-3、PDGFR-β、Flt-3和c-KIT,对应的IC50值分别为90 nM、20 nM、57 nM、59 nM和68 nM。Sorafenib 可诱导autophagy、apoptosis并激活ferroptosis,Sorafenib 具有抗肿瘤活性。 |
-
J Hepatocell Carcinoma, 2026, 13:572919
-
Mol Cancer, 2025, 24(1):34
-
Nat Commun, 2025, 16(1):509
|
|
| S1040 |
Sorafenib Tosylate (BAY 43-9006)
|
Sorafenib tosylate是Raf-1和B-Raf的多重激酶抑制剂,无细胞试验中IC50分别为6 nM和22 nM。Sorafenib 还可抑制VEGFR-2、VEGFR-3、PDGFR-β、Flt-3和c-KIT,对应的IC50值分别为90 nM、20 nM、57 nM、59 nM和68 nM。Sorafenib Tosylate 可诱导autophagy、apoptosis并激活ferroptosis,并具有抗肿瘤活性。 |
-
Int J Oncol, 2025, 67(3)72
-
Nature, 2024, 629(8013):927-936
-
Cell Mol Life Sci, 2024, 81(1):238
|
|
| S7781 |
Sunitinib (SU-11248)
|
Sunitinib是一种多靶点 RTK 抑制剂,以VEGFR2(Flk-1)和PDGFRβ为靶点,IC50为80 nM 和 2 nM,对c-Kit也有抑制作用。Sunitinib 还是 IRE1α 的自磷酸化活性的剂量依赖性抑制剂。 舒尼替尼可诱导自噬和细胞凋亡。 |
-
Cancer Cell, 2025, 43(4):776-796.e14
-
Mol Cancer, 2025, 24(1):179
-
J Exp Clin Cancer Res, 2025, 44(1):290
|

|
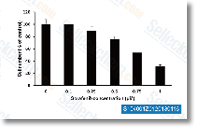
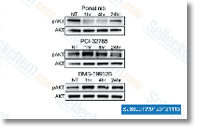
| S1490 |
Ponatinib (AP24534)
|
Ponatinib是一种新型有效的多靶点抑制剂,在无细胞试验中作用于Abl,PDGFRα,VEGFR2,FGFR1和Src,IC50分别为0.37 nM,1.1 nM,1.5 nM,2.2 nM和5.4 nM。Ponatinib (AP24534)可抑制自噬。 |
-
J Cell Mol Med, 2026, 30(4):e71053
-
Cancers (Basel), 2026, 18(7)1082
-
Nat Commun, 2025, 16(1):471
|
|
| S1178 |
Regorafenib (BAY 73-4506)
|
Regorafenib是一个多靶点抑制剂,作用于VEGFR1,VEGFR2,VEGFR3,PDGFR-β,Kit (c-Kit),RET (c-RET)和Raf-1,在无细胞试验中IC50分别是13 nM,4.2 nM,46 nM,22 nM,7 nM,1.5 nM和2.5 nM。Regorafenib 可诱导自噬。 |
-
Nat Commun, 2025, 16(1):509
-
Cell Death Differ, 2025, 10.1038/s41418-025-01536-1
-
J Nanobiotechnology, 2025, 24(1):64
|
|
| S1005 |
Axitinib (AG-013736)
|
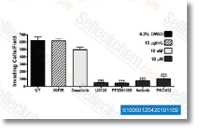
Axitinib是一种多靶点抑制剂,作用于 VEGFR1,VEGFR2,VEGFR3,PDGFRβ和c-Kit,在猪主动脉内皮细胞中IC50分别为0.1 nM,0.2 nM,0.1-0.3 nM,1.6 nM和1.7 nM。 |
-
Pharmacol Res, 2026, 225:108112
-
Development, 2026, 153(16)dev205344
-
J Exp Zool B Mol Dev Evol, 2026, 346(1):7-19
|
|
| S1029 |
CC-5013 (Lenalidomide)
|
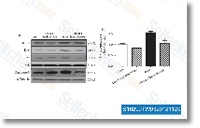
Lenalidomide是一种TNF-α分泌抑制剂,在PBMCs中IC50为13 nM。Lenalidomide是 ubiquitin E3 ligase cereblon (CRBN) 的配体,它通过CRBN-CRL4泛素连接酶引起两个淋巴样转录因子IKZF1和IKZF3的选择性泛素化和降解。Lenalidomide 可促进 cleaved caspase-3 的表达、抑制 VEGF 的表达并诱导凋亡。 |
-
Signal Transduct Target Ther, 2025, 10(1):29
-
Nat Commun, 2025, 16(1):3800
-
Cell Rep Med, 2025, S2666-3791(25)00102-8
|
|
| S1042 |
Sunitinib (SU11248) Malate
|
Sunitinib malate是一种多靶点RTK抑制剂,作用于VEGFR2 (Flk-1)和 PDGFRβ,在无细胞试验中IC50分别为80 nM 和2 nM,也会抑制c-Kit的活性。Sunitinib Malate 可有效地抑制 Ire1α 的自身磷酸化。Sunitinib Malate 可增加 death receptor 和 线粒体依赖的凋亡 mitochondrial-dependent apoptosis。 |
-
Nat Commun, 2025, 16(1):7853
-
CNS Neurosci Ther, 2025, 31(12):e70696
-
Sci Rep, 2025, 15(1):35889
|
|
| S1119 |
Cabozantinib (XL184)
|
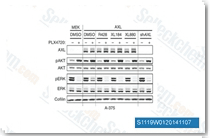
Cabozantinib是一种有效的VEGFR2抑制剂,在无细胞试验中IC50为0.035 nM,也能有效抑制c-Met、 Ret、 Kit、Flt-1/3/4、Tie2和AXL,IC50分别为1.3 nM,4 nM,4.6 nM,12 nM/11.3 nM/6 nM,14.3 nM 和 7 nM。Cabozantinib 在结肠癌细胞中可通过AKT/GSK-3β/NF-κB信号通路诱导PUMA依赖的凋亡。 |
-
Cell Reports Medicine, September 20, 2022, 100659
-
Redox Biology, October 21, 2023, 102945
-
Hepatology Communications, November 8, 2023, e0313
|
|
| S1010 |
BIBF 1120 (Nintedanib)
|
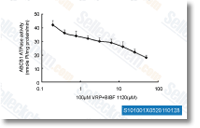
Nintedanib尼达尼布是一种有效的三重血管激酶抑制剂,作用于VEGFR1/2/3, FGFR1/2/3和PDGFRα/β,在无细胞试验中IC50分别为34 nM/13 nM/13 nM, 69 nM/37 nM/108 nM和59 nM/65 nM。Phase 3。 |
-
Cancer Biology & Medicine, November 19, 2025, 20250275
-
Thorax, February 18, 2026, thorax-2025-223325
-
Drug Design, Development and Therapy, February 18, 2022, 397-411
|
|
| S1164 |
E7080 (Lenvatinib)
|
Lenvatinib是一种多靶点抑制剂,无细胞试验中,作用于VEGFR2(KDR)/VEGFR3(Flt-4)最有效,IC50为4 nM/5.2 nM,对VEGFR1/Flt-1作用效果稍弱,作用于VEGFR2/3比作用于FGFR1, PDGFRα/β选择性高10倍左右。Lenvatinib (E7080) 也是FGFR1-4、PDGFR、Kit (c-Kit)和RET (c-RET)的抑制剂,并具有强效的抗肿瘤活性。Phase 3。 |
-
Int J Biol Sci, 2026, 22(7):3617-3634
-
Cancer Sci, 2026, 117(1):257-271
-
J Hepatocell Carcinoma, 2026, 13:572919
|
|
| S1264 |
PD173074
|
PD173074 是一种有效的FGFR1抑制剂,在无细胞试验中IC50约为25 nM,也能抑制VEGFR2,IC50为100-200 nM,作用于FGFR1比作用于PDGFR和c-Src选择性高1000倍左右。PD173074 可在胃癌细胞中抑制增殖并促进凋亡。 |
-
Cell Rep, 2025, 44(10):116310
-
Stem Cell Reports, 2025, 20(10):102640
-
Commun Biol, 2025, 8(1):125
|

|
| S1046 |
Vandetanib (ZD6474)
|
Vandetanib是一种有效的 VEGFR2 抑制剂,在无细胞试验中IC50为40 nM。同时,也抑制VEGFR3和EGFR,IC50分别为110 nM 和500 nM。对PDGFRβ, Flt-1, Tie-2 和FGFR1作用效果不大,IC50为 1.1-3.6 μM, 对MEK, CDK2, c-Kit, erbB2, FAK, PDK1, Akt 和 IGF-1R几乎没有作用效果,IC50>10 μM。Vandetanib (ZD6474) 可增加凋亡并通过提高 reactive oxygen species (ROS) 的水平来诱导自噬。 |
-
Frontiers in Pharmacology, May 10, 2024, 15:1345070
-
Molecular Cancer Therapeutics, March 15, 2016, 503-11
-
Oncotarget, June 14, 2016, 36101-36114
|
|
| S4001 |
Cabozantinib S-malate
|
Cabozantinib malate (XL184)是Cabozantinib的苹果酸盐,是有效的VEGFR2抑制剂,IC50为0.035 nM,也抑制c-Met, RET (c-RET), Kit (c-Kit), Flt-1/3/4, Tie2和AXL,无细胞试验中IC50分别为1.3 nM, 4 nM, 4.6 nM, 12 nM/11.3 nM/6 nM, 14.3 nM和7 nM。Cabozantinib malate (XL184)可诱导细胞凋亡。 |
-
Scientific Reports, 2025, 35889
-
Sci Rep, 2025, 15(1):35889
-
bioRxiv, 2025, 2025.08.15.670608
|
|
| S1111 |
Foretinib (GSK1363089, XL880)
|
Foretinib是一种ATP竞争性的HGFR和VEGFR抑制剂,对Met (c-Met)和KDR作用最强,在无细胞试验中IC50分别为0.4 nM和0.9 nM。对Ron, Flt-1/3/4, Kit (c-Kit), PDGFRα/β和Tie-2作用效果稍弱,对FGFR1和EGFR几乎没有抑制活性。Phase 2。 |
-
J Microbiol, 2025, 63(2):e2409001
-
Int J Mol Sci, 2023, 24(1)757
-
Biol Open, 2023, 12(8)bio059994
|

|
| S3012 |
Pazopanib (GW786034)
|
Pazopanib (GW786034) 是一种新型多靶点的VEGFR1,VEGFR2,VEGFR3,PDGFR,FGFR,c-Kit 和 c-Fms/CSF1R抑制剂,无细胞试验中IC50分别为10 nM,30 nM,47 nM,84 nM,74 nM,140 nM 和 146 nM。Pazopanib 可诱导 cathepsin B 的活化和自噬。 |
-
Nucleic Acids Res, 2025, 53(4)gkaf107
-
Commun Biol, 2025, 8(1):1185
-
Sci Rep, 2025, 15(1):35889
|

|
| S1017 |
Cediranib (AZD2171)
|
Cediranib (AZD2171, NSC-732208)是一种高效的VEGFR(KDR)抑制剂,IC50为<1 nM,同时也抑制Flt1/4,IC50为5 nM/≤3 nM,此外对c-Kit和PDGFRβ也具有相似的抑制活性,对VEGFR的选择性比PDGFR-α, CSF-1R和Flt3分别高36倍, 110倍 和1000倍以上。Cediranib (AZD2171)可诱导自噬小体累积。Phase 3。 |
-
Front Oncol, 2024, 14:1302850
-
Cell Oncol (Dordr), 2023, 46(2):391-407
-
Biochem Pharmacol, 2023, 214:115681
|
|
| S1035 |
Pazopanib HCl
|
Pazopanib HCl是一种新型多靶点抑制剂,作用于VEGFR1,VEGFR2,VEGFR3,PDGFR,FGFR,c-Kit和c-fms,在无细胞试验中IC50分别是10 nM,30 nM,47 nM,84 nM,74 nM,140 nM和146 nM。Pazopanib 可诱导自噬II型细胞死亡。 |
-
The Journal of Clinical Investigation, 2025, e185220
-
Nucleic Acids Research, 2025, gkaf107
-
iScience, 2024, 27(10):110862
|

|
| S8064 |
Midostaurin (PKC412)
|
Midostaurin 是一种多靶点激酶抑制剂,作用于PKCα/β/γ, Syk, Flk-1, Akt, PKA, c-Kit, c-Fgr, c-Src, FLT3, PDFRβ和VEGFR1/2,IC50为80-500 nM。 |
-
Cancer Res, 2026, 86(7):1724-1738
-
Blood Adv, 2025, bloodadvances.2024015427
-
Onco Targets Ther, 2025, 18:489-501
|
|
| S1101 |
Vatalanib (PTK787) 2HCl
|
Vatalanib 2HCl (PTK787, ZK 222584, cpg-79787) 是一种VEGFR2/KDR抑制剂,无细胞试验中IC50为37 nM,对VEGFR1/Flt-1作用稍弱,是VEGFR3/Flt-4抑制作用的18倍。Phase 3。 |
-
Nat Commun, 2025, 16(1):7287
-
Environ Health Perspect, 2024, 132(5):57001
-
J Cancer, 2024, 15(9):2448-2459
|

|
| S1018 |
Dovitinib (TKI-258)
|
Dovitinib (TKI-258, CHIR-258)是一种多靶点的RTK抑制剂,在无细胞试验中对III型(FLT3/c-Kit)作用最强,IC50为1 nM/2 nM,同时也作用于IV类(FGFR1/3)和V类(VEGFR1-4) RTKs,IC50为8-13 nM,但对InsR,EGFR,c-Met,EphA2,Tie2,IGF-1R和HER2作用较弱。Phase 4。 |
-
Spectrochim Acta A Mol Biomol Spectrosc, 2025, 343:126602
-
Biomed Pharmacother, 2024, 180:117533
-
Sci Rep, 2023, 13(1):20223
|

|
| S5077 |
Regorafenib (BAY-734506) Monohydrate
|
Regorafenib (BAY-734506, Fluoro-sorafenib, Resihance, Stivarga, regorafaenib monohydrate) Monohydrate 是一种新型口服的多重激酶抑制剂,对VEGFR1、小鼠VEGFR2、小鼠VEGFR3、PDGFR-β、Kit (c-Kit)、RET (c-RET)、RAF-1、B-RAF和B-RAF(V600E)的IC50分别为13, 4.2, 46, 22, 7, 1.5, 2.5, 28, 19 nM。 |
-
Nature, 2024, 629(8011):450-457
-
iScience, 2024, 27(10):110862
-
STAR Protoc, 2024, 5(2):103090
|
|
| S1207 |
Tivozanib
|
Tivozanib是一种有效的,选择性VEGFR抑制剂,作用于VEGFR1/2/3时,IC50分别为0.21 nM/0.16 nM/0.24 nM,也抑制PDGFR和c-Kit,作用于FGFR-1, Flt3, c-Met EGFR和IGF-1R活性较弱。Phase 3。 |
-
Acta Pharmacol Sin, 2026, 10.1038/s41401-026-01791-z
-
bioRxiv, 2025, 2025.01.24.634732
-
Nat Commun, 2024, 15(1):4758
|

|
| S8401 |
Erdafitinib (JNJ-42756493)
|
Erdafitinib是有效的、具有选择性和口服生物活性的泛成纤维细胞生长因子受体FGFR抑制剂,具有潜在的抗肿瘤活性。Erdafitinib 也能结合RET (c-RET)、CSF-1R、PDGFR-α/PDGFR-β、FLT4、Kit (c-Kit)和VEGFR-2并可诱导细胞凋亡。 |
-
Commun Biol, 2025, 8(1):394
-
Int J Mol Sci, 2025, 26(8)3525
-
J Clin Invest, 2024, 134(2)e169241
|

|
| S1003 |
Linifanib (ABT-869)
|
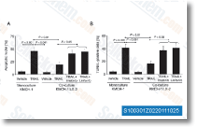
Linifanib (ABT-869, AL39324, RG3635)是一种新型有效的ATP竞争性VEGFR/PDGFR抑制剂,作用于KDR,CSF-1R,Flt-1/3和PDGFRβ,其IC50分别为4 nM,3 nM,3 nM/4 nM和66 nM,对突变激酶依赖性癌细胞(即FLT3)最有效。Linifanib (ABT-869) 可诱导自噬和凋亡。Phase 3。 |
-
Research in Pharmaceutical Sciences, 2025 Oct 20, 734-745
-
Biomed Pharmacother, 2024, 180:117533
-
Cell Death Discovery, 2023 Feb 10, 57
|
|
| S2842 |
SAR131675
|
SAR131675是一种VEGFR3抑制剂,无细胞试验中IC50/Ki为23 nM/12 nM,作用于VEGFR3比作用于VEGFR1/2选择性高50和10倍,对Akt1, CDKs, PLK1, EGFR, IGF-1R, c-Met, Flt2等几乎没有作用活性。 |
-
Cell Death Discov, 2025, 11(1):320
-
Zool Res, 2025, 46(6):1317-1325
-
Cell Signal, 2025, 130:111675
|

|
| S5240 |
Lenvatinib Mesylate
|
Lenvatinib Mesylate是一种合成、具有口服活性的 tyrosine kinase 的抑制剂,可抑制血管内皮生长因子受体(VEGFR1-3),成纤维细胞生长因子受体(FGFR1-4),血小板衍生生长因子受体(PDGFRα),干细胞因子受体(Kit (c-Kit)),并在转染过程中重新排列(RET (c-RET))。甲磺酸来伐替尼具有潜在的抗肿瘤活性。 |
-
Nat Commun, 2024, 15(1):1754
-
iScience, 2024, 27(10):110862
-
J Hepatocell Carcinoma, 2023, 10:697-712
|
|
| S7765 |
Dovitinib (TKI258) Lactate monohydrate
|
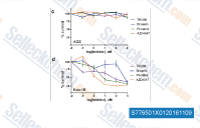
Dovitinib (TKI258) Lactate monohydrate是Dovitinib的乳酸盐,Dovitinib是一种多靶点的PTK抑制剂,主要对第III类(FLT3/c-Kit)发挥作用,IC50为1 nM/2 nM,对第IV 类(FGFR1/3)和第V 类(VEGFR1-4) RTKs也有效,IC50为8-13 nM,对InsR,EGFR,c-Met,EphA2,Tie2,IGFR1 和HER2作用较差。Phase 4。
|
-
Mol Cell, 2021, S1097-2765(21)00507-4
-
Acta Neuropathol, 2021, 10.1007/s00401-021-02327-x
-
Cancer Res, 2021, canres.1780.2020
|
|
| S5248 |
Apatinib (YN968D1)
|
Apatinib (Rivoceranib, YN968D1) 是一种有效的VEGF信号通路的抑制剂,其对VEGFR-2、Ret、c-Kit和c-Src的IC50值分别为1 nM、13 nM、429 nM和530 nM。Apatinib可诱导自噬和凋亡。 |
-
Anticancer Research, October 2019, 5461-5471
-
Frontiers in Oncology, November 18, 2021, 739139
-
Ann Dermatol, 2025, 37(4):228-240
|
|
| S2221 |
Apatinib (YN968D1) mesylate
|
Apatinib mesylate (YN968D1, Rivoceranib)是一种有效的VEGF信号通路的抑制剂,其对VEGFR-2、Ret、c-Kit和c-Src的IC50值分别为1 nM、13 nM、429 nM和530 nM。Apatinib mesylate 可诱导自噬和凋亡。 |
-
Oncol Res, 2025, 33(6):1459-1472
-
Clin Respir J, 2024, 18(2):e13738
-
Discov Oncol, 2024, 15(1):709
|
|
| S7667 |
SU 5402
|
SU5402是一种有效的多靶点受体激酶抑制剂,对VEGFR2,FGFR1,和PDGF-Rβ的IC50分别为20 nM,30 nM,和510 nM。
|
-
Exploration (Beijing), 2026, 6(2):70160
-
Int J Mol Sci, 2025, 26(8)3536
-
Basic Clin Pharmacol Toxicol, 2025, 136(5):e70022
|

|
| S2161 |
RAF265 (CHIR-265)
|
RAF265 (CHIR-265)是一种有效的选择性C-Raf/B-Raf/B-Raf V600E抑制剂,IC50为3-60 nM,对VEGFR2磷酸化表现出有效的抑制作用,无细胞试验中EC50为30 nM。RAF265 (CHIR-265)可诱导细胞周期阻滞和凋亡。Phase 2。 |
-
bioRxiv, May 3, 2025, nan
-
Molecular Cancer Therapeutics, December 2015, 2700-11
-
bioRxiv, 2025, 2025.04.29.651188
|

|
| S1363 |
Ki8751
|
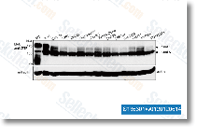
Ki8751是一种有效的,选择性的VEGFR2抑制剂,IC50为0.9 nM,作用于VEGFR2比作用于c-Kit, PDGFRα和FGFR-2选择性高40倍以上,对EGFR, HGFR和InsR没有抑制活性。 |
-
Theranostics, January 1, 2026, Not specified
-
American Journal of Physiology-Renal Physiology, January 1, 2023, F106-F123
-
Elife, 2025, 13RP98612
|
|
| S4947 |
Regorafenib Hydrochloride
|
Regorafenib (Stivarga, BAY 73-4506) Hydrochloride 是一种多靶点抑制剂,对 VEGFR1、Murine VEGFR2/3、PDGFRβ、Kit (c-Kit)、RET (c-RET) 和 Raf-1 的IC50值分别为13 nM、4.2 nM/46 nM、22 nM、7 nM、1.5 nM和2.5 nM。 |
-
Cancers (Basel), 2023, 15(18)4477
-
J Clin Med, 2023, 12(23)7267
-
J Hematol Oncol, 2022, 15(1):109
|
|
| S2845 |
Semaxanib (SU5416)
|
Semaxanib (SU5416, semaxinib)是一种有效的,选择性的VEGFR(Flk-1/KDR)抑制剂,IC50为1.23 μM,作用于VEGFR比作用于PDGFRβ选择性高20倍,对EGFR, InsR和FGFR没有作用活性。Phase 3。Semaxanib (SU5416)可用于诱导低氧模型动物疾病模型。 |
-
Clin Sci (Lond), 2025, 139(10)CS20255922
-
bioRxiv, 2025, 2025.01.24.634732
-
Nat Commun, 2024, 15(1):4758
|

|
| S2896 |
ZM 323881 HCl
|
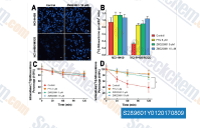
ZM 323881 HCl是一种有效的,选择性的VEGFR2抑制剂,IC50为<2 nM,对VEGFR1, PDGFRβ, FGFR1, EGFR和ErbB2几乎没有活性。 |
-
Molecular Pain, January 15, 2022, 17448069221075891
-
European Journal of Medicinal Chemistry, December 5, 2018, 193-206
-
iScience, August 7, 2023, 107548
|
|
| S5242 |
Cediranib Maleate
|
Cediranib Maleate (AZD2171) is the maleate salt of Cediranib, which is a potent inhibitor of VEGFR with IC50 of <1 nM and also inhibits Flt1/4 with IC50 of 5 nM/≤3 nM. |
-
J Exp Clin Cancer Res, 2024, 43(1):56
-
Journal of Experimental & Clinical Cancer Research, 2024, 56
-
International Journal of Molecular Sciences, 2023, 8037
|
|
| S5234 |
Nintedanib Ethanesulfonate Salt
|
Nintedanib (Intedanib, BIBF 1120) is a small molecule tyrosine-kinase inhibitor with IC50 of 34 nM/13 nM/13 nM, 69 nM/37 nM/108 nM and 59 nM/65 nM for VEGFR1/2/3, FGFR1/2/3 and PDGFRα/β, respectively. |
-
Cell, 2026, S0092-8674(26)00223-0
-
iScience, 2024, 27(10):110862
-
Advanced Science, 2023, e2300383
|
|
| S1486 |
AEE788 (NVP-AEE788)
|
AEE788 (NVP-AEE788)是一种有效的EGFR和HER2/ErbB2抑制剂,IC50分别为2 nM和6 nM,对VEGFR2/KDR, c-Abl, c-Src,和Flt-1作用效果稍弱,对Ins-R, IGF-1R, PKCα和CDK1没有抑制作用。Phase 1/2。 |
-
Cell Rep Med, 2022, 3(1):100492
-
Genome Med, 2020, 18;12(1):17
-
Oncotarget, 2019, 10(68):7185-7197
|

|
| S2897 |
ZM 306416
|
ZM 306416 (CB 676475) 是一种VEGFR(Flt和KDR)抑制剂,作用于VEGFR1,IC50为0.33 μM,也抑制EGFR,IC50为<10 nM。 |
-
PLoS One, September 1, 2016, e0161332
-
Journal of Dental Sciences, October 2022, 1471-1479
-
Glia, November 2018, 2503-2513
|

|
| S7057 |
LY2874455
|
LY2874455是一个泛FGFR的抑制剂,对FGFR1,FGFR2, FGFR3, 和FGFR4的IC50值分别为2.8 nM, 2.6 nM, 6.4 nM,以及6 nM。并且也能抑制VEGFR2的活性,IC50为7 nM。Phase 1。
|
-
Cancer Drug Resist, 2025, 8:45
-
Cancer Discov, 2023, 13(9):1998-2011
-
Cell Rep, 2022, 39(12):110994
|

|
| S4686 |
Vitamin E
|
Vitamin E (D-alpha-Tocopherol) 是一种脂溶性的维生素,具有有效的抗氧化特性。它是有效的过氧化氢自由基清除剂并在很多组织中,非竞争性地抑制环氧酶活性。同时通过抑制VEGF基因转录,抑制血管生成和肿瘤休眠。 |
-
Nutrients, September 14, 2022, 14(18):3791
-
eLife, June 28, 2024, 13:RP94169
-
Cell, January 9, 2025, 188(1):157-174.e22
|
|
| S1032 |
Motesanib Diphosphate (AMG-706)
|
Motesanib Diphosphate (AMG-706)是一种有效的ATP竞争性的VEGFR1/2/3抑制剂,IC50分别为2 nM/3 nM/6 nM;对Kit (c-Kit)具有相似的抑制活性,对VEGFR选择性比PDGFR和Ret高10倍。Phase 3。 |
-
Cell, April 5, 2018, 515-528.e17
-
Front Cell Dev Biol, 2022, 10:836179
-
J Pharm Sci, 2022, 111(8):2180-2190
|

|
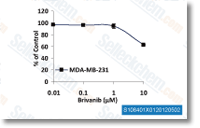
| S1084 |
Brivanib (BMS-540215)
|
Brivanib (BMS-540215)是一种ATP竞争性的VEGFR2抑制剂,IC50为25 nM,对VEGFR-1和FGFR-1抑制作用适中,但比作用于PDGFR-β效果强240多倍。Phase 3。 |
-
Molecular Pharmaceutics, October 21, 2019, 4436-4450
-
Molecular Pharmaceutics, November 4, 2019, 4436-4450
-
Experimental & Molecular Medicine, November 16, 2022, 2007-2021
|
|
| S1361 |
MGCD-265 analog
|
MGCD-265是一种有效的,多靶点,及ATP竞争性的c-Met和VEGFR1/2/3抑制剂,IC50分别为1 nM, 3 nM/3 nM/4 nM,也抑制Ron和Tie2。Phase 1/2。 |
-
Cell & Bioscience, 2024, 109
-
Cancer Cell, 2022, S1535-6108(22)00312-9
-
Cell Rep Med, 2022, 3(1):100492
|

|
| S1181 |
ENMD-2076
|
ENMD-2076 选择性地作用于 Aurora A 和 Flt3,IC50分别为14 nM和1.86 nM,作用于Aurora A比作用于Aurora B选择性高25倍,对RET、SRC、NTRK1/TRKA、CSF1R/FMS、VEGFR2/KDR、FGFR和PDGFRα作用效果稍弱。ENMD-2076 可抑制各种人类实体瘤和造血癌细胞系的生长,其IC50值为0.025至0.7μM,可诱导凋亡和G2/M期停滞。Phase 2。 |
-
Int J Biol Macromol, 2025, 292:139119
-
Cell, 2021, 184(2):334-351.e20
-
Cancer Discovery, 2021, 2544-2563
|

|
| S1171 |
CYC116
|
CYC116是一种有效的Aurora A/B抑制剂,Ki为8.0 nM/9.2 nM,对VEGFR2(Ki为44 nM)作用稍弱,比作用于CDKs效果强50倍,对PKA, Akt/PKB, PKC没有活性,对GSK-3α/β, CK2, Plk1和SAPK2A.没有抑制效果。Phase 1。 |
-
Scientific Reports, April 2, 2024, 7739
-
Proceedings of the National Academy of Sciences, April 12, 2022, e2122512119
-
Scientific Reports, 2024, 7739
|

|
| S5793 |
Motesanib (AMG-706)
|
Motesanib (AMG-706)是一种具有口服生物活性的受体酪氨酸激酶 (receptor tyrosine kinase)抑制剂,对VEGFR1, VEGFR2, VEGFR3, Kit, PDGFR和Ret的IC50值分别为2、3、6、8、84和59 nM。 |
-
Signal Transduct Target Ther, 2022, 7(1):147
-
Front Cell Dev Biol, 2022, 10:836179
-
J Pharm Sci, 2022, 111(8):2180-2190
|
|
| S8573 |
Sitravatinib (MGCD516)
|
Sitravatinib (MGCD516, MG-516)是一种新型的、靶向多种参与调节S180肉瘤细胞生长的RTKs的小分子抑制剂,包括c-Kit, PDGFRβ, PDGFRα, c-Met和Axl。 |
-
Translational Cancer Research, October 2019, 2425-2438
-
Cancer Research, October 15, 2025, 3983-3998
-
Neuro-Oncology, May 2025, 1372–1384
|
|
| S1220 |
OSI-930
|
OSI-930是一种有效的Kit (c-Kit), KDR和CSF-1R抑制剂,IC50分别为80 nM, 9 nM和15 nM,对Flt-1, c-Raf和Lck具有适度的抑制活性,对PDGFRα/β, Flt-3和Abl抑制活性较弱。Phase 1。 |
-
Nat Commun, 2024, 15(1):4739
-
Cell Rep, 2019, 28(9):2331-2344
-
Nat Biomed Eng, 2018, 2(8):578-588
|
|
| S2859 |
Golvatinib (E7050)
|
Golvatinib (E7050)是c-Met和VEGFR-2双重抑制剂,IC50分别为14 nM和16 nM,不抑制bFGF刺激的HUVEC生长(浓度高达1000 nM)。Phase 1/2。 |
-
Int J Mol Sci, 2023, 24(11)9606
-
International Journal of Molecular Sciences, 2023, 9606
-
International Journal of Molecular Sciences, 2022, 14884
|

|
| S2622 |
PP121
|
PP121是一种作用于PDGFR, Hck, mTOR, VEGFR2, Src和Abl的多靶点抑制剂,IC50分别为2 nM, 8 nM, 10 nM, 12 nM, 14 nM和18 nM,也抑制DNA-PK,IC50为60 nM。 |
-
PLOS One, October 21, 2016, e0164895
-
Cancer Research, November 01, 2015, 4548-4559
-
Life Sci Alliance, 2021, 4(2)e202000882
|

|
| S2366 |
Taxifolin (Dihydroquercetin)
|
Taxifolin(Dihydroquercetin)是一种二氢黄酮醇类,是一种黄酮类化合物。是I型VEGFR2 kinase的抑制剂。 |
-
J Virol, 2025, e0209524.
-
Foods, 2025, 14(24)4314
-
Foods, 2025, 4314
|
|
| S1557 |
KRN 633
|
KRN 633是一种ATP竞争性的VEGFR1/2/3抑制剂,IC50为170 nM/160 nM/125 nM,微弱抑制PDGFR-α/β和c-Kit,对细胞中FGFR-1, EGFR和c-Met的磷酸化没有抑制作用。 |
-
Aust Dent J, 2020, 10.1111/adj.12752
-
J Med Invest, 2017, 64(3.4):262-265
-
Oncotarget, 2016, 7(5):5985-99
|
|
| S5667 |
Fruquintinib (HMPL-013)
|
Fruquintinib is a small molecule inhibitor with strong potency and high selectivity against VEGFR family. It inhibits VEGFR 1, 2, 3, with IC50 values of 33 nM, 35 nM and 0.5 nM, respectively and shows only weak inhibition of RET, FGFR-1 and c-kit kinases. |
-
Sci Adv, 2025, 11(20):eads1631
-
EMBO J, 2024, 43(8):1519-1544
-
iScience, 2024, 27(10):110862
|
|
| S2231 |
Telatinib
|
Telatinib (BAY 57-9352) 是一种有效的VEGFR2/3, c-Kit和PDGFRα抑制剂,IC50分别为6 nM/4 nM, 1 nM和15 nM。Phase 2。 |
-
Sci Adv, 2022, 8(6):eabg9455
-
Leiden University Scholarly Publications, 2020, N/A
-
Cell Stem Cell, 2019, 24(4):654-669
|
|
| S8721 |
PDGFR inhibitor 1
|
PDGFR inhibitor 1是一种具有口服活性的Kit (c-Kit)和PDGFR抑制剂,具有潜在的抗肿瘤活性。它还能抑制好几种其他激酶,包括VEGFR2、TIE2、PDGFR-beta和CSF1R,因此进而抑制肿瘤细胞的生长。 |
-
Res Sq, 2025, rs.3.rs-7032881
-
bioRxiv, 2025, 2025.06.11.659120
-
eLife, 2023, e79840
|
|
| S7258 |
SKLB1002
|
SKLB1002 是一种与ATP竞争的强效VEGFR2抑制剂,其IC50为32 nM. |
-
Sci Rep, 2025, 15(1):8177
-
Sci Adv, 2024, 10(20):eadl0633
-
Dis Model Mech, 2023, 16(5)dmm049571
|

|
| S2201 |
BMS-794833
|
BMS-794833是一种有效的,ATP竞争性Met (c-Met)/VEGFR2抑制剂,IC50为1.7 nM/15 nM,也抑制Ron, Axl和Flt3,IC50低于3 nM;是BMS-817378的前体药物。 Phase 1。 |
-
Frontiers in Oncology, August 20, 2021, 686765
-
Cell Stem Cell, 2025, 32(11):1671-1690.e13
-
Cancer Res, 2021, canres.1428.2021
|
|
| S8189 |
BAW2881 (NVP-BAW2881)
|
BAW2881 (NVP-BAW2881)是一种新型的VEGFR酪氨酸激酶抑制剂。在1.0-4.3 nM浓度下能有效地抑制VEGFR1-3;在45-72 nM的浓度下,抑制PDGFRβ, c-Kit和RET (c-RET)。 |
-
J Transl Med, 2025, 23(1):985
-
mediaTUM, 2022, 158
-
JCI Insight, 2021, 143285
|
|
| S5272 |
Toceranib phosphate
|
Toceranib phosphate (Palladia, SU11654), the phosphate salt of toceranib, is a selective inhibitor of the tyrosine kinase activity of several members of the split kinase RTK family, including Flk-1/KDR, PDGFR, and Kit with Ki values of 6 nM and 5 nM for Flk-1/KDR and PDGFRβ, respectively. |
-
Frontiers in Veterinary Science, May 21, 2024, 1392728
-
Journal of Pharmacology and Experimental Therapeutics, February 15, 2024, 774-787
-
Liver International, April 2023, 928-944
|
|
| S7003 |
AZD2932
|
AZD2932 是一种有效的多靶点蛋白酪氨酸激酶抑制剂,对VEGFR-2,PDGFRβ,Flt-3,和 c-Kit 的 IC50 分别为 8 nM,4 nM,7 nM,和 9 nM。
|
-
Cancer Research, 2021, 747-762
-
Cancer Res, 2020, canres.1992.2020
-
Molecules, 2020, 8;25(9) pii: E2220
|
|
| S0504 |
SU14813
|
SU14813 (SU 014813)是一种多受体 tyrosine kinase 的抑制剂,对于 VEGFR2、VEGFR1、PDGFRβ 和 Kit (c-Kit) 的IC50值分别为50 nM,2 nM,4 nM和15 nM。SU14813 具有强大的抗血管生成和抗肿瘤活性。 |
-
Cancer Cell, 2022, S1535-6108(22)00312-9
-
Adv Sci (Weinh), 2021, e2101848
-
Advanced Science, 2021, e2101848
|
|
| S0278 |
SU5614
|
SU5614 (Chloro-SU5416, Chloro-Semaxanib) 是一种小分子 receptor tyrosine kinases (RTK) 的抑制剂,对 VEGFR-2、c-kit 以及野生型和突变型 FLT3 均有抑制作用。SU5614 可减少细胞增殖并诱导凋亡。 |
-
Frontiers in Cell and Developmental Biology, January 26, 2022, 797047
-
Advanced Science, August 7, 2021, e2101848
-
Am J Pathol, 2024, S0002-9440(24)00326-2
|
|
| S0765 |
MAZ51
|
MAZ51 是一种选择性的 vascular endothelial growth factor receptor (VEGFR)-3 (Flt-4) tyrosine kinase 的有效抑制剂。MAZ51 通过 Akt/GSK3β 的磷酸化和 RhoA 的激活来诱导神经胶质瘤细胞的细胞圆缩和G2/M细胞周期停滞。 MAZ51 可抑制多种非VEGFR-3表达肿瘤细胞的增殖并诱导其凋亡。 |
-
Sci Rep, 2025, 15(1):1283
-
International Immunopharmacology, 2025, 114443
-
Human Molecular Genetics, 2014, 6553-6566
|
|
| S6412 |
Altiratinib
|
Altiratinib (DCC-2701) 是有效的TRK, Met (c-Met), TIE2和VEGFR2激酶抑制剂,对TRKA, B和C的IC50分别为0.9 nM, 4.6 nM和0.8 nM。它抑制Met (c-Met)和Met (c-Met)突变体的IC50范围为0.3-6 nM。 |
-
J Microbiol, 2025, 63(2):e2409001
-
Theranostics, 2021, 11(20):9918-9936
-
Cold Spring Harb Mol Case Stud, 2021, 7(5)a006109
|
|
| S1138 |
Brivanib Alaninate (BMS-582664)
|
Brivanib Alaninate (BMS-582664)是BMS-540215的前体药物,是一种ATP竞争性的VEGFR2抑制剂,IC50为25 nM。 |
-
Int J Oncol, 2014, 44(3):959-69
-
Assay Drug Dev Technol, 2014, 12(9-10):514-26
|
|
| S2018 |
ENMD-2076 L-(+)-Tartaric acid
|
ENMD-2076 L-(+)-Tartaric acid 是ENMD-2076的酒石酸,选择性作用于Aurora A和VEGFR(Flt3),IC50分别为14 nM和1.86 nM,作用于Aurora A比作用于Aurora B选择性高25倍,对VEGFR2/KDR和VEGFR3, FGFR1和FGFR2和PDGFRα作用效果稍弱。Phase 2。 |
-
Veterinary and Comparative Oncology, 2016 Mar, 14(1):13-27
-
International Journal of Oncology, 2015 Jul, 47(1):122-32
|
|
| S6535 |
SU1498
|
SU1498是KDR抑制剂,IC50为0.7 μM,可在内皮细胞中刺激磷酸化ERK1/2的累积。 |
-
Cell Metab, 2020, 5;31(5):892-908 e11
-
Biol Open, 2018,
|
|
| S7688 |
Ki20227
|
Ki20227 是一种口服活性的 c-Fms tyrosine kinase(CSF1R) 的高选择性抑制剂,对 c-Fms、vascular endothelial growth factor receptor-2 (KDR/VEGFR-2)、stem cell factor receptor (c-Kit) 和 platelet-derived growth factor receptor beta (PDGFRβ) 的IC50值为2 nM、12 nM、451 nM和217 nM。 |
-
Cancer Immunol Immunother, 2024, 73(5):76
-
Arteriosclerosis, Thrombosis, and Vascular Biology, 2023, 1063-1078
|
|
| S6526 |
SKLB 610
|
SKLB-610是一种多靶点的酪氨酸激酶 (tyrosine kinases)抑制剂。它对VEGFR2的抑制作用最为有效,是FGFR2和PDGFR的弱抑制剂。 |
-
Biomed Pharmacother, 2022, 149:112922
-
Biomedicine & Pharmacotherapy, 2022, 112922
-
International Journal of Molecular Sciences, 2022, 7853
|
|
| S6514 |
SU5408
|
SU5408 (VEGFR2 Kinase Inhibitor I)是一种有效、选择性 VEGFR2 Kinase抑制剂,IC50为70 nM。 |
-
Transl Oncol, 2022, 25:101516
-
Translational Oncology, 2022, 101516
-
J Dent Sci, 2022, 17(4):1471-1479
|
|
| S0487 |
Sulfatinib
|
Sulfatinib是一种有效的、具有高度选择性的 tyrosine kinase 的抑制剂,对于 VEGFR1、VEGFR2、VEGFR3、FGFR1 和 CSF1R 的IC50值分别为2 nM、24 nM、1 nM、15 nM和4 nM。Sulfatinib 在针对晚期NET患者的实验中显示出令人鼓舞的抗肿瘤活性和可控毒性。 |
-
International Journal of Biological Sciences, 2025, 7043-7062
-
Frontiers in Oncology, 2023, 1158857
-
Front Oncol, 2023, 13:1158857
|
|
| S0290 |
SU5204
|
SU5204 是一种酪氨酸激酶抑制剂,对 FLK-1 (VEGFR-2) 和 HER2 的IC50值分别为4 μM和51.5 μM。 |
-
Front Pharmacol, 2021, 12:804327
|
|
| S2211 |
AG-13958
|
AG-13958 (AG-013958)是VEGFR酪氨酸激酶抑制剂,在临床开发中发现ST给药可治疗与年龄相关性黄斑变性(AMD)的脉络膜新生血管生成。 |
-
iScience, 2023, 107548
|
|
| S7647 |
Lucitanib (E3810) hydrochloride
|
Lucitanib (E-3810, AL3810) hydrochloride 是 Vascular endothelial growth factor receptor (VEGFR) 和 Fibroblast growth factor receptor (FGFR) 的双重抑制剂。 Lucitanib hydrochloride (E-3810, AL3810) 有效且选择性地抑制 VEGFR1、VEGFR2、VEGFR3、FGFR1 和 FGFR2,IC50值分别为7 nM、25 nM、10 nM、17.5 nM和82.5 nM。 |
-
Sci Rep, 2023, 13(1):20223
|
|
| S6520 |
WHI-P180
|
WHI-P180是多种激酶的抑制剂,对RET (c-RET)和KDR的IC50值分别为4.5 nM和66 nM。 |
|
|
| S6543 |
ZD-4190
|
ZD-4190是VEGF RTK抑制剂,对KDR和Flt-1的IC50值分别为29 ± 4 nM和708 ± 63 nM。 |
|
|
| S8188 |
BFH772
|
BFH772是一种新型的、有效的、可口服的VEGFR2抑制剂,IC50为3 nM。 |
|
|
| S0291 |
SU5208
|
SU5208(3-[(Thien-2-yl)methylene]-2-indolinone)是血管内皮生长因子受体2(vascular endothelial growth factor receptor-2, VEGFR2)的抑制剂。 |
|
|
| E2397 |
hVEGF-IN-1
|
hVEGF-IN-1是一种喹唑啉衍生物,可特异性结合到核糖体内部进入位点A(internal ribosome entry site A, IRES-A)的G-rich序列,Kd为0.928 μM,从而破坏G -四复合物结构;并可降低VEGF-A蛋白表达,从而阻碍肿瘤细胞迁移,抑制肿瘤生长。 |
|
|
| S8882 |
ODM-203
|
ODM-203是FGFR 和 VEGFR的双重抑制剂,对重组FGFR1,FGFR2,FGFR3,FGFR4,VEGFR1,VEGFR2和VEGFR3的ic50分别为11 nM,16 nM,6 nM,35 nM,26 nM,9 nM,5 nM。 |
|
|
| S0763 |
Tyrphostin AG1433
|
Tyrphostin AG1433 (AG1433, SU1433)是一种选择性血小板衍生生长因子受体β (platelet-derived growth factor receptor β, PDGFRβ)和血管内皮生长因子受体2 (vascular endothelial growth factor receptor 2, VEGFR-2, Flk-1/KDR) 抑制剂,IC50分别为5.0 μM和9.3 μM。 |
|
|
| E0499 |
4SC-203
|
4SC-203(SC71710)是一种具有潜在抗肿瘤活性的多激酶抑制剂。4SC-203选择性抑制FMS相关酪氨酸激酶3(FMS-related tyrosine kinase 3, FLT3/STK1)、FLT3的突变形式和血管内皮生长因子受体(vascular endothelial growth factor receptors, VEGFRs)。
|
|
|
| E2656 |
R1530
|
R1530是一种有效的、口服活性的、双重作用的有丝分裂/血管生成抑制剂,可抑制血管内皮生长因子受体2 (vascular endothelial growth factor receptor 2, VEGFR2)和成纤维细胞生长因子受体1(fibroblast growth factor receptors 1, FGFR1),IC50分别为10 nM和28 nM。 |
|
|
| S9621 |
Donafenib (Sorafenib D3)
|
Donafenib (Sorafenib D3, Bay 43-9006 D3, CM-4307)是氘标记的Sorafenib。Sorafenib 是一种多激酶的抑制剂,对 Raf-1、mVEGFR-2、mVEGFR-3 和 B-RAF 的抑制作用对应的IC50值分别为6 nM、15 nM、20 nM和22 nM。 |
|
|
| E1078 |
Emvododstat (PTC299)
|
Emvododstat(PTC299)是致病性VEGF和双氢乳清酸酯脱氢酶(dihydroorotate dehydrogenase, DHODH)的转录后抑制剂,可抑制VEGFAmRNA翻译和异常细胞增殖。在HeLa细胞中,PTC299抑制缺氧诱导的VEGFA蛋白的产生,EC50为1.64 nM。 |
|
|
| S5607 |
4,4'-Bis(4-aminophenoxy)biphenyl
|
4,4'-Bis(4-aminophenoxy)biphenyl is a monomer for polyimide production. |
|
|
| E0616 |
Chiauranib
|
Chiauranib (CS2164)能够选择性抑制多种激酶靶标,包括aurora B激酶(aurora B kinase, AURKB)、集落刺激因子1受体(colony-stimulating factor 1 receptor, CSF1R)和血管内皮生长因子受体(vascular endothelial growth factor receptor, VEGFR)/血小板衍生生长因子受体(platelet-derived growth factor receptor, PDGFR)/c -Kit,从而抑制肿瘤细胞的快速增殖,增强抗肿瘤免疫,抑制肿瘤血管生成,达到抗肿瘤功效。 |
|
|
| S6870 |
Ningetinib
|
Ningetinib (CT-053, DE-120, CT053PTSA)是一种有效的、可口服的 tyrosine kinase,对 c-Met、VEGFR2 和 Axl 的IC50值分别为6.7 nM、1.9 nM和 <1.0 nM。Ningetinib 表现出了抗肿瘤的活性。 |
|
|
| S6843 |
Vorolanib
|
Vorolanib (X-82,CM082)是一种口服的 vascular endothelial growth factor receptor (VEGFR) 和 platelet-derived growth factor receptor (PDGFR) 的多激酶双重抑制剂,并具有抗血管生成和抗肿瘤的活性。 |
|
|
| S6764 |
Pamufetinib (TAS-115)
|
Pamufetinib (TAS-115) 是一种高效的 c-Met 和 VEGFR 双重抑制剂,对重组 VEGFR2 和重组 MET 的 IC50 分别为 30 nM 和 32 nM。 |
|
|
| S6939 |
Nastorazepide
|
Nastorazepide (Z-360)是一种胆囊收缩素-2/胃泌素受体(cholecystokinin-2/gastrin receptor, CCK2/胃泌素受体)拮抗剂,与吉西他滨联合使用可延长胰腺癌原位异种移植小鼠的存活率并降低吉西他滨诱导的血管内皮生长因子(vascular endothelial growth factor, VEGF)表达。 |
|
|
| E3379 |
Cassia seed Extract
|
Cassia seed Extract是从决明(Cassia toraLinn)的种子中提取的,具有影响多种生物学过程和途径的潜力,包括对平滑肌细胞增殖、炎症反应、肿瘤坏死因子(TNF)信号通路的正调控 、血管内皮生长因子(VEGF)信号通路和花生四烯酸(ARA)代谢。 |
|
|
| S0377 |
CS-2660 (JNJ-38158471)
|
CS-2660 (JNJ-38158471) 是一种耐受性良好的、口服有效的、高选择性的 VEGFR-2 抑制剂,IC50值为40 nM。CS-2660(JNJ-38158471)还抑制紧密相关的酪氨酸激酶,如 Ret (c-RET) 和 Kit (c-Kit),IC50为180 nM和500 nM,但无明显活性 (>1 microM) 针对VEGFR-1和VEGFR-3。 |
|
|
| E0534 |
WAY-340935
|
WAY-340935 (VEGFR2-IN-2)可以抑制VEGFR2的功能,其对H460细胞系的抗增殖活性部分是通过与VEGFR蛋白的相互作用而产生的。 |
|
|
| S9402 |
(20R)-Protopanaxadiol
|
(20R)-Protopanaxadiol, isolated from the roots of Panax ginseng, has protective effect on myocardial ischemia. |
|
|
| E3414 |
Semen litchi Extract
|
Semen litchi Extract是从荔枝Litchi chinensis中提取的,具有抗肝癌作用,并通过靶向VEGFR抑制肝细胞癌血管生成。 |
|
|
| E4914 |
Cabozantinib hydrochloride
|
Cabozantinib hydrochloride(XL184, BMS-907351 hydrochloride) 是一种有效的 c-MET 和 VEGFR2 小分子激酶抑制剂,IC50 分别为 1.3 nM、0.035 nM。它还抑制 RET、KIT、AXL、Tie2 和 FLT3,IC50 分别为 5.2 nM、4.6 nM、7 nM、14.3 nM、11.3 nM。它可以成为抑制 MET 和 VEGFR 信号失调的癌症中的肿瘤血管生成和转移的有希望的药物。 |
|
|
| E0207 |
Chebulinic acid
|
Chebulinic acid,一种从榄仁树果实中分离的酚类化合物,是一种新型流感病毒神经氨酸酶(Influenza viral neuraminidase)抑制剂。Chebulinic acid 也是一种抗血管生成剂,通过抑制 VEGF 起作用。Chebulinic acid 具有抗肿瘤活性。 |
|
|
| E3774 |
Chlorella Pyrenoidosa Extract
|
Chlorella Pyrenoidosa Extract是从小球藻Chlorella Pyrenoidosa中提取的,对VEGF诱导的新生血管有明显的抗血管生成活性和抗增殖活性。 |
|
|
| E4694 |
EVT801
|
EVT801 是一种强效且选择性的 VEGFR-3 抑制剂,IC50 为 11 nM。它具有强效的抗肿瘤作用,是一种有前途的抗淋巴管生成药物,适用于 VEGFR-3 阳性肿瘤患者。 |
|
|
| S6808 |
SU5205
|
SU5205是 VEGF receptor 2 (VEGFR2/FLK-1) 的抑制剂,IC50为9.6 µM。 |
|
|
| S6809 |
SU5214
|
SU5214是 VEGF receptor 2 (VEGFR2/FLK-1) 和 EGFR 的抑制剂,对应的IC50值分别为14.8 µM 和 36.7 µM。 |
|
|
| E1435 |
Tinengotinib
|
Tinengotinib (TT-00420) 是一种新型多激酶抑制剂,可强力抑制 Aurora A/B,IC50 值分别为 1.2/3.3 nM。 它还对 FGFR1/2/3、VEGFR1、JAK1/2 和 CSF1R 表现出有效的抑制活性,可用于治疗三阴性乳腺癌 (TNBC) 。 |
|
|
| E0142 |
XL092
|
XL092 (JUN04542) 是多种 RTK 的 ATP 竞争性抑制剂,包括 MET、VEGFR2、AXL 和 MER,在基于细胞的测定中 IC50 值分别为 15 nM、1.6 nM、3.4 nM 和 7.2 nM。 |
|
|
| S7397 |
Sorafenib (BAY 43-9006)
|
Sorafenib是Raf-1和B-Raf的多重激酶抑制剂,无细胞试验中IC50分别为6 nM和22 nM。Sorafenib 还可抑制VEGFR-2、VEGFR-3、PDGFR-β、Flt-3和c-KIT,对应的IC50值分别为90 nM、20 nM、57 nM、59 nM和68 nM。Sorafenib 可诱导autophagy、apoptosis并激活ferroptosis,Sorafenib 具有抗肿瘤活性。 |
- J Hepatocell Carcinoma, 2026, 13:572919
- Mol Cancer, 2025, 24(1):34
- Nat Commun, 2025, 16(1):509
|

|
| S1040 |
Sorafenib Tosylate (BAY 43-9006)
|
Sorafenib tosylate是Raf-1和B-Raf的多重激酶抑制剂,无细胞试验中IC50分别为6 nM和22 nM。Sorafenib 还可抑制VEGFR-2、VEGFR-3、PDGFR-β、Flt-3和c-KIT,对应的IC50值分别为90 nM、20 nM、57 nM、59 nM和68 nM。Sorafenib Tosylate 可诱导autophagy、apoptosis并激活ferroptosis,并具有抗肿瘤活性。 |
- Int J Oncol, 2025, 67(3)72
- Nature, 2024, 629(8013):927-936
- Cell Mol Life Sci, 2024, 81(1):238
|

|
| S7781 |
Sunitinib (SU-11248)
|
Sunitinib是一种多靶点 RTK 抑制剂,以VEGFR2(Flk-1)和PDGFRβ为靶点,IC50为80 nM 和 2 nM,对c-Kit也有抑制作用。Sunitinib 还是 IRE1α 的自磷酸化活性的剂量依赖性抑制剂。 舒尼替尼可诱导自噬和细胞凋亡。 |
- Cancer Cell, 2025, 43(4):776-796.e14
- Mol Cancer, 2025, 24(1):179
- J Exp Clin Cancer Res, 2025, 44(1):290
|

|
| S1490 |
Ponatinib (AP24534)
|
Ponatinib是一种新型有效的多靶点抑制剂,在无细胞试验中作用于Abl,PDGFRα,VEGFR2,FGFR1和Src,IC50分别为0.37 nM,1.1 nM,1.5 nM,2.2 nM和5.4 nM。Ponatinib (AP24534)可抑制自噬。 |
- J Cell Mol Med, 2026, 30(4):e71053
- Cancers (Basel), 2026, 18(7)1082
- Nat Commun, 2025, 16(1):471
|

|
| S1178 |
Regorafenib (BAY 73-4506)
|
Regorafenib是一个多靶点抑制剂,作用于VEGFR1,VEGFR2,VEGFR3,PDGFR-β,Kit (c-Kit),RET (c-RET)和Raf-1,在无细胞试验中IC50分别是13 nM,4.2 nM,46 nM,22 nM,7 nM,1.5 nM和2.5 nM。Regorafenib 可诱导自噬。 |
- Nat Commun, 2025, 16(1):509
- Cell Death Differ, 2025, 10.1038/s41418-025-01536-1
- J Nanobiotechnology, 2025, 24(1):64
|

|
| S1005 |
Axitinib (AG-013736)
|
Axitinib是一种多靶点抑制剂,作用于 VEGFR1,VEGFR2,VEGFR3,PDGFRβ和c-Kit,在猪主动脉内皮细胞中IC50分别为0.1 nM,0.2 nM,0.1-0.3 nM,1.6 nM和1.7 nM。 |
- Pharmacol Res, 2026, 225:108112
- Development, 2026, 153(16)dev205344
- J Exp Zool B Mol Dev Evol, 2026, 346(1):7-19
|

|
| S1029 |
CC-5013 (Lenalidomide)
|
Lenalidomide是一种TNF-α分泌抑制剂,在PBMCs中IC50为13 nM。Lenalidomide是 ubiquitin E3 ligase cereblon (CRBN) 的配体,它通过CRBN-CRL4泛素连接酶引起两个淋巴样转录因子IKZF1和IKZF3的选择性泛素化和降解。Lenalidomide 可促进 cleaved caspase-3 的表达、抑制 VEGF 的表达并诱导凋亡。 |
- Signal Transduct Target Ther, 2025, 10(1):29
- Nat Commun, 2025, 16(1):3800
- Cell Rep Med, 2025, S2666-3791(25)00102-8
|

|
| S1042 |
Sunitinib (SU11248) Malate
|
Sunitinib malate是一种多靶点RTK抑制剂,作用于VEGFR2 (Flk-1)和 PDGFRβ,在无细胞试验中IC50分别为80 nM 和2 nM,也会抑制c-Kit的活性。Sunitinib Malate 可有效地抑制 Ire1α 的自身磷酸化。Sunitinib Malate 可增加 death receptor 和 线粒体依赖的凋亡 mitochondrial-dependent apoptosis。 |
- Nat Commun, 2025, 16(1):7853
- CNS Neurosci Ther, 2025, 31(12):e70696
- Sci Rep, 2025, 15(1):35889
|
|
| S1119 |
Cabozantinib (XL184)
|
Cabozantinib是一种有效的VEGFR2抑制剂,在无细胞试验中IC50为0.035 nM,也能有效抑制c-Met、 Ret、 Kit、Flt-1/3/4、Tie2和AXL,IC50分别为1.3 nM,4 nM,4.6 nM,12 nM/11.3 nM/6 nM,14.3 nM 和 7 nM。Cabozantinib 在结肠癌细胞中可通过AKT/GSK-3β/NF-κB信号通路诱导PUMA依赖的凋亡。 |
- Cell Reports Medicine, September 20, 2022, 100659
- Redox Biology, October 21, 2023, 102945
- Hepatology Communications, November 8, 2023, e0313
|

|
| S1010 |
BIBF 1120 (Nintedanib)
|
Nintedanib尼达尼布是一种有效的三重血管激酶抑制剂,作用于VEGFR1/2/3, FGFR1/2/3和PDGFRα/β,在无细胞试验中IC50分别为34 nM/13 nM/13 nM, 69 nM/37 nM/108 nM和59 nM/65 nM。Phase 3。 |
- Cancer Biology & Medicine, November 19, 2025, 20250275
- Thorax, February 18, 2026, thorax-2025-223325
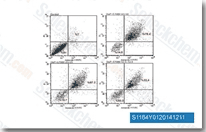
- Drug Design, Development and Therapy, February 18, 2022, 397-411
|

|
| S1164 |
E7080 (Lenvatinib)
|
Lenvatinib是一种多靶点抑制剂,无细胞试验中,作用于VEGFR2(KDR)/VEGFR3(Flt-4)最有效,IC50为4 nM/5.2 nM,对VEGFR1/Flt-1作用效果稍弱,作用于VEGFR2/3比作用于FGFR1, PDGFRα/β选择性高10倍左右。Lenvatinib (E7080) 也是FGFR1-4、PDGFR、Kit (c-Kit)和RET (c-RET)的抑制剂,并具有强效的抗肿瘤活性。Phase 3。 |
- Int J Biol Sci, 2026, 22(7):3617-3634
- Cancer Sci, 2026, 117(1):257-271
- J Hepatocell Carcinoma, 2026, 13:572919
|

|
| S1264 |
PD173074
|
PD173074 是一种有效的FGFR1抑制剂,在无细胞试验中IC50约为25 nM,也能抑制VEGFR2,IC50为100-200 nM,作用于FGFR1比作用于PDGFR和c-Src选择性高1000倍左右。PD173074 可在胃癌细胞中抑制增殖并促进凋亡。 |
- Cell Rep, 2025, 44(10):116310
- Stem Cell Reports, 2025, 20(10):102640
- Commun Biol, 2025, 8(1):125
|

|
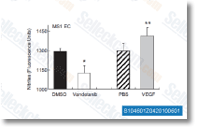
| S1046 |
Vandetanib (ZD6474)
|
Vandetanib是一种有效的 VEGFR2 抑制剂,在无细胞试验中IC50为40 nM。同时,也抑制VEGFR3和EGFR,IC50分别为110 nM 和500 nM。对PDGFRβ, Flt-1, Tie-2 和FGFR1作用效果不大,IC50为 1.1-3.6 μM, 对MEK, CDK2, c-Kit, erbB2, FAK, PDK1, Akt 和 IGF-1R几乎没有作用效果,IC50>10 μM。Vandetanib (ZD6474) 可增加凋亡并通过提高 reactive oxygen species (ROS) 的水平来诱导自噬。 |
- Frontiers in Pharmacology, May 10, 2024, 15:1345070
- Molecular Cancer Therapeutics, March 15, 2016, 503-11
- Oncotarget, June 14, 2016, 36101-36114
|

|
| S4001 |
Cabozantinib S-malate
|
Cabozantinib malate (XL184)是Cabozantinib的苹果酸盐,是有效的VEGFR2抑制剂,IC50为0.035 nM,也抑制c-Met, RET (c-RET), Kit (c-Kit), Flt-1/3/4, Tie2和AXL,无细胞试验中IC50分别为1.3 nM, 4 nM, 4.6 nM, 12 nM/11.3 nM/6 nM, 14.3 nM和7 nM。Cabozantinib malate (XL184)可诱导细胞凋亡。 |
- Scientific Reports, 2025, 35889
- Sci Rep, 2025, 15(1):35889
- bioRxiv, 2025, 2025.08.15.670608
|
|
| S1111 |
Foretinib (GSK1363089, XL880)
|
Foretinib是一种ATP竞争性的HGFR和VEGFR抑制剂,对Met (c-Met)和KDR作用最强,在无细胞试验中IC50分别为0.4 nM和0.9 nM。对Ron, Flt-1/3/4, Kit (c-Kit), PDGFRα/β和Tie-2作用效果稍弱,对FGFR1和EGFR几乎没有抑制活性。Phase 2。 |
- J Microbiol, 2025, 63(2):e2409001
- Int J Mol Sci, 2023, 24(1)757
- Biol Open, 2023, 12(8)bio059994
|

|
| S3012 |
Pazopanib (GW786034)
|
Pazopanib (GW786034) 是一种新型多靶点的VEGFR1,VEGFR2,VEGFR3,PDGFR,FGFR,c-Kit 和 c-Fms/CSF1R抑制剂,无细胞试验中IC50分别为10 nM,30 nM,47 nM,84 nM,74 nM,140 nM 和 146 nM。Pazopanib 可诱导 cathepsin B 的活化和自噬。 |
- Nucleic Acids Res, 2025, 53(4)gkaf107
- Commun Biol, 2025, 8(1):1185
- Sci Rep, 2025, 15(1):35889
|

|
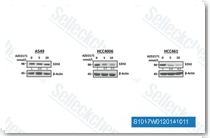
| S1017 |
Cediranib (AZD2171)
|
Cediranib (AZD2171, NSC-732208)是一种高效的VEGFR(KDR)抑制剂,IC50为<1 nM,同时也抑制Flt1/4,IC50为5 nM/≤3 nM,此外对c-Kit和PDGFRβ也具有相似的抑制活性,对VEGFR的选择性比PDGFR-α, CSF-1R和Flt3分别高36倍, 110倍 和1000倍以上。Cediranib (AZD2171)可诱导自噬小体累积。Phase 3。 |
- Front Oncol, 2024, 14:1302850
- Cell Oncol (Dordr), 2023, 46(2):391-407
- Biochem Pharmacol, 2023, 214:115681
|

|
| S1035 |
Pazopanib HCl
|
Pazopanib HCl是一种新型多靶点抑制剂,作用于VEGFR1,VEGFR2,VEGFR3,PDGFR,FGFR,c-Kit和c-fms,在无细胞试验中IC50分别是10 nM,30 nM,47 nM,84 nM,74 nM,140 nM和146 nM。Pazopanib 可诱导自噬II型细胞死亡。 |
- The Journal of Clinical Investigation, 2025, e185220
- Nucleic Acids Research, 2025, gkaf107
- iScience, 2024, 27(10):110862
|

|
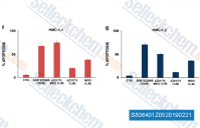
| S8064 |
Midostaurin (PKC412)
|
Midostaurin 是一种多靶点激酶抑制剂,作用于PKCα/β/γ, Syk, Flk-1, Akt, PKA, c-Kit, c-Fgr, c-Src, FLT3, PDFRβ和VEGFR1/2,IC50为80-500 nM。 |
- Cancer Res, 2026, 86(7):1724-1738
- Blood Adv, 2025, bloodadvances.2024015427
- Onco Targets Ther, 2025, 18:489-501
|

|
| S1101 |
Vatalanib (PTK787) 2HCl
|
Vatalanib 2HCl (PTK787, ZK 222584, cpg-79787) 是一种VEGFR2/KDR抑制剂,无细胞试验中IC50为37 nM,对VEGFR1/Flt-1作用稍弱,是VEGFR3/Flt-4抑制作用的18倍。Phase 3。 |
- Nat Commun, 2025, 16(1):7287
- Environ Health Perspect, 2024, 132(5):57001
- J Cancer, 2024, 15(9):2448-2459
|

|
| S1018 |
Dovitinib (TKI-258)
|
Dovitinib (TKI-258, CHIR-258)是一种多靶点的RTK抑制剂,在无细胞试验中对III型(FLT3/c-Kit)作用最强,IC50为1 nM/2 nM,同时也作用于IV类(FGFR1/3)和V类(VEGFR1-4) RTKs,IC50为8-13 nM,但对InsR,EGFR,c-Met,EphA2,Tie2,IGF-1R和HER2作用较弱。Phase 4。 |
- Spectrochim Acta A Mol Biomol Spectrosc, 2025, 343:126602
- Biomed Pharmacother, 2024, 180:117533
- Sci Rep, 2023, 13(1):20223
|

|
| S5077 |
Regorafenib (BAY-734506) Monohydrate
|
Regorafenib (BAY-734506, Fluoro-sorafenib, Resihance, Stivarga, regorafaenib monohydrate) Monohydrate 是一种新型口服的多重激酶抑制剂,对VEGFR1、小鼠VEGFR2、小鼠VEGFR3、PDGFR-β、Kit (c-Kit)、RET (c-RET)、RAF-1、B-RAF和B-RAF(V600E)的IC50分别为13, 4.2, 46, 22, 7, 1.5, 2.5, 28, 19 nM。 |
- Nature, 2024, 629(8011):450-457
- iScience, 2024, 27(10):110862
- STAR Protoc, 2024, 5(2):103090
|
|
| S1207 |
Tivozanib
|
Tivozanib是一种有效的,选择性VEGFR抑制剂,作用于VEGFR1/2/3时,IC50分别为0.21 nM/0.16 nM/0.24 nM,也抑制PDGFR和c-Kit,作用于FGFR-1, Flt3, c-Met EGFR和IGF-1R活性较弱。Phase 3。 |
- Acta Pharmacol Sin, 2026, 10.1038/s41401-026-01791-z
- bioRxiv, 2025, 2025.01.24.634732
- Nat Commun, 2024, 15(1):4758
|

|
| S8401 |
Erdafitinib (JNJ-42756493)
|
Erdafitinib是有效的、具有选择性和口服生物活性的泛成纤维细胞生长因子受体FGFR抑制剂,具有潜在的抗肿瘤活性。Erdafitinib 也能结合RET (c-RET)、CSF-1R、PDGFR-α/PDGFR-β、FLT4、Kit (c-Kit)和VEGFR-2并可诱导细胞凋亡。 |
- Commun Biol, 2025, 8(1):394
- Int J Mol Sci, 2025, 26(8)3525
- J Clin Invest, 2024, 134(2)e169241
|

|
| S1003 |
Linifanib (ABT-869)
|
Linifanib (ABT-869, AL39324, RG3635)是一种新型有效的ATP竞争性VEGFR/PDGFR抑制剂,作用于KDR,CSF-1R,Flt-1/3和PDGFRβ,其IC50分别为4 nM,3 nM,3 nM/4 nM和66 nM,对突变激酶依赖性癌细胞(即FLT3)最有效。Linifanib (ABT-869) 可诱导自噬和凋亡。Phase 3。 |
- Research in Pharmaceutical Sciences, 2025 Oct 20, 734-745
- Biomed Pharmacother, 2024, 180:117533
- Cell Death Discovery, 2023 Feb 10, 57
|

|
| S2842 |
SAR131675
|
SAR131675是一种VEGFR3抑制剂,无细胞试验中IC50/Ki为23 nM/12 nM,作用于VEGFR3比作用于VEGFR1/2选择性高50和10倍,对Akt1, CDKs, PLK1, EGFR, IGF-1R, c-Met, Flt2等几乎没有作用活性。 |
- Cell Death Discov, 2025, 11(1):320
- Zool Res, 2025, 46(6):1317-1325
- Cell Signal, 2025, 130:111675
|

|
| S5240 |
Lenvatinib Mesylate
|
Lenvatinib Mesylate是一种合成、具有口服活性的 tyrosine kinase 的抑制剂,可抑制血管内皮生长因子受体(VEGFR1-3),成纤维细胞生长因子受体(FGFR1-4),血小板衍生生长因子受体(PDGFRα),干细胞因子受体(Kit (c-Kit)),并在转染过程中重新排列(RET (c-RET))。甲磺酸来伐替尼具有潜在的抗肿瘤活性。 |
- Nat Commun, 2024, 15(1):1754
- iScience, 2024, 27(10):110862
- J Hepatocell Carcinoma, 2023, 10:697-712
|
|
| S7765 |
Dovitinib (TKI258) Lactate monohydrate
|
Dovitinib (TKI258) Lactate monohydrate是Dovitinib的乳酸盐,Dovitinib是一种多靶点的PTK抑制剂,主要对第III类(FLT3/c-Kit)发挥作用,IC50为1 nM/2 nM,对第IV 类(FGFR1/3)和第V 类(VEGFR1-4) RTKs也有效,IC50为8-13 nM,对InsR,EGFR,c-Met,EphA2,Tie2,IGFR1 和HER2作用较差。Phase 4。
|
- Mol Cell, 2021, S1097-2765(21)00507-4
- Acta Neuropathol, 2021, 10.1007/s00401-021-02327-x
- Cancer Res, 2021, canres.1780.2020
|

|
| S5248 |
Apatinib (YN968D1)
|
Apatinib (Rivoceranib, YN968D1) 是一种有效的VEGF信号通路的抑制剂,其对VEGFR-2、Ret、c-Kit和c-Src的IC50值分别为1 nM、13 nM、429 nM和530 nM。Apatinib可诱导自噬和凋亡。 |
- Anticancer Research, October 2019, 5461-5471
- Frontiers in Oncology, November 18, 2021, 739139
- Ann Dermatol, 2025, 37(4):228-240
|
|
| S2221 |
Apatinib (YN968D1) mesylate
|
Apatinib mesylate (YN968D1, Rivoceranib)是一种有效的VEGF信号通路的抑制剂,其对VEGFR-2、Ret、c-Kit和c-Src的IC50值分别为1 nM、13 nM、429 nM和530 nM。Apatinib mesylate 可诱导自噬和凋亡。 |
- Oncol Res, 2025, 33(6):1459-1472
- Clin Respir J, 2024, 18(2):e13738
- Discov Oncol, 2024, 15(1):709
|
|
| S7667 |
SU 5402
|
SU5402是一种有效的多靶点受体激酶抑制剂,对VEGFR2,FGFR1,和PDGF-Rβ的IC50分别为20 nM,30 nM,和510 nM。
|
- Exploration (Beijing), 2026, 6(2):70160
- Int J Mol Sci, 2025, 26(8)3536
- Basic Clin Pharmacol Toxicol, 2025, 136(5):e70022
|

|
| S2161 |
RAF265 (CHIR-265)
|
RAF265 (CHIR-265)是一种有效的选择性C-Raf/B-Raf/B-Raf V600E抑制剂,IC50为3-60 nM,对VEGFR2磷酸化表现出有效的抑制作用,无细胞试验中EC50为30 nM。RAF265 (CHIR-265)可诱导细胞周期阻滞和凋亡。Phase 2。 |
- bioRxiv, May 3, 2025, nan
- Molecular Cancer Therapeutics, December 2015, 2700-11
- bioRxiv, 2025, 2025.04.29.651188
|

|
| S1363 |
Ki8751
|
Ki8751是一种有效的,选择性的VEGFR2抑制剂,IC50为0.9 nM,作用于VEGFR2比作用于c-Kit, PDGFRα和FGFR-2选择性高40倍以上,对EGFR, HGFR和InsR没有抑制活性。 |
- Theranostics, January 1, 2026, Not specified
- American Journal of Physiology-Renal Physiology, January 1, 2023, F106-F123
- Elife, 2025, 13RP98612
|

|
| S4947 |
Regorafenib Hydrochloride
|
Regorafenib (Stivarga, BAY 73-4506) Hydrochloride 是一种多靶点抑制剂,对 VEGFR1、Murine VEGFR2/3、PDGFRβ、Kit (c-Kit)、RET (c-RET) 和 Raf-1 的IC50值分别为13 nM、4.2 nM/46 nM、22 nM、7 nM、1.5 nM和2.5 nM。 |
- Cancers (Basel), 2023, 15(18)4477
- J Clin Med, 2023, 12(23)7267
- J Hematol Oncol, 2022, 15(1):109
|
|
| S2845 |
Semaxanib (SU5416)
|
Semaxanib (SU5416, semaxinib)是一种有效的,选择性的VEGFR(Flk-1/KDR)抑制剂,IC50为1.23 μM,作用于VEGFR比作用于PDGFRβ选择性高20倍,对EGFR, InsR和FGFR没有作用活性。Phase 3。Semaxanib (SU5416)可用于诱导低氧模型动物疾病模型。 |
- Clin Sci (Lond), 2025, 139(10)CS20255922
- bioRxiv, 2025, 2025.01.24.634732
- Nat Commun, 2024, 15(1):4758
|

|
| S2896 |
ZM 323881 HCl
|
ZM 323881 HCl是一种有效的,选择性的VEGFR2抑制剂,IC50为<2 nM,对VEGFR1, PDGFRβ, FGFR1, EGFR和ErbB2几乎没有活性。 |
- Molecular Pain, January 15, 2022, 17448069221075891
- European Journal of Medicinal Chemistry, December 5, 2018, 193-206
- iScience, August 7, 2023, 107548
|

|
| S5242 |
Cediranib Maleate
|
Cediranib Maleate (AZD2171) is the maleate salt of Cediranib, which is a potent inhibitor of VEGFR with IC50 of <1 nM and also inhibits Flt1/4 with IC50 of 5 nM/≤3 nM. |
- J Exp Clin Cancer Res, 2024, 43(1):56
- Journal of Experimental & Clinical Cancer Research, 2024, 56
- International Journal of Molecular Sciences, 2023, 8037
|
|
| S5234 |
Nintedanib Ethanesulfonate Salt
|
Nintedanib (Intedanib, BIBF 1120) is a small molecule tyrosine-kinase inhibitor with IC50 of 34 nM/13 nM/13 nM, 69 nM/37 nM/108 nM and 59 nM/65 nM for VEGFR1/2/3, FGFR1/2/3 and PDGFRα/β, respectively. |
- Cell, 2026, S0092-8674(26)00223-0
- iScience, 2024, 27(10):110862
- Advanced Science, 2023, e2300383
|
|
| S1486 |
AEE788 (NVP-AEE788)
|
AEE788 (NVP-AEE788)是一种有效的EGFR和HER2/ErbB2抑制剂,IC50分别为2 nM和6 nM,对VEGFR2/KDR, c-Abl, c-Src,和Flt-1作用效果稍弱,对Ins-R, IGF-1R, PKCα和CDK1没有抑制作用。Phase 1/2。 |
- Cell Rep Med, 2022, 3(1):100492
- Genome Med, 2020, 18;12(1):17
- Oncotarget, 2019, 10(68):7185-7197
|

|
| S2897 |
ZM 306416
|
ZM 306416 (CB 676475) 是一种VEGFR(Flt和KDR)抑制剂,作用于VEGFR1,IC50为0.33 μM,也抑制EGFR,IC50为<10 nM。 |
- PLoS One, September 1, 2016, e0161332
- Journal of Dental Sciences, October 2022, 1471-1479
- Glia, November 2018, 2503-2513
|

|
| S7057 |
LY2874455
|
LY2874455是一个泛FGFR的抑制剂,对FGFR1,FGFR2, FGFR3, 和FGFR4的IC50值分别为2.8 nM, 2.6 nM, 6.4 nM,以及6 nM。并且也能抑制VEGFR2的活性,IC50为7 nM。Phase 1。
|
- Cancer Drug Resist, 2025, 8:45
- Cancer Discov, 2023, 13(9):1998-2011
- Cell Rep, 2022, 39(12):110994
|

|
| S4686 |
Vitamin E
|
Vitamin E (D-alpha-Tocopherol) 是一种脂溶性的维生素,具有有效的抗氧化特性。它是有效的过氧化氢自由基清除剂并在很多组织中,非竞争性地抑制环氧酶活性。同时通过抑制VEGF基因转录,抑制血管生成和肿瘤休眠。 |
- Nutrients, September 14, 2022, 14(18):3791
- eLife, June 28, 2024, 13:RP94169
- Cell, January 9, 2025, 188(1):157-174.e22
|
|
| S1032 |
Motesanib Diphosphate (AMG-706)
|
Motesanib Diphosphate (AMG-706)是一种有效的ATP竞争性的VEGFR1/2/3抑制剂,IC50分别为2 nM/3 nM/6 nM;对Kit (c-Kit)具有相似的抑制活性,对VEGFR选择性比PDGFR和Ret高10倍。Phase 3。 |
- Cell, April 5, 2018, 515-528.e17
- Front Cell Dev Biol, 2022, 10:836179
- J Pharm Sci, 2022, 111(8):2180-2190
|

|
| S1084 |
Brivanib (BMS-540215)
|
Brivanib (BMS-540215)是一种ATP竞争性的VEGFR2抑制剂,IC50为25 nM,对VEGFR-1和FGFR-1抑制作用适中,但比作用于PDGFR-β效果强240多倍。Phase 3。 |
- Molecular Pharmaceutics, October 21, 2019, 4436-4450
- Molecular Pharmaceutics, November 4, 2019, 4436-4450
- Experimental & Molecular Medicine, November 16, 2022, 2007-2021
|

|
| S1361 |
MGCD-265 analog
|
MGCD-265是一种有效的,多靶点,及ATP竞争性的c-Met和VEGFR1/2/3抑制剂,IC50分别为1 nM, 3 nM/3 nM/4 nM,也抑制Ron和Tie2。Phase 1/2。 |
- Cell & Bioscience, 2024, 109
- Cancer Cell, 2022, S1535-6108(22)00312-9
- Cell Rep Med, 2022, 3(1):100492
|

|
| S1181 |
ENMD-2076
|
ENMD-2076 选择性地作用于 Aurora A 和 Flt3,IC50分别为14 nM和1.86 nM,作用于Aurora A比作用于Aurora B选择性高25倍,对RET、SRC、NTRK1/TRKA、CSF1R/FMS、VEGFR2/KDR、FGFR和PDGFRα作用效果稍弱。ENMD-2076 可抑制各种人类实体瘤和造血癌细胞系的生长,其IC50值为0.025至0.7μM,可诱导凋亡和G2/M期停滞。Phase 2。 |
- Int J Biol Macromol, 2025, 292:139119
- Cell, 2021, 184(2):334-351.e20
- Cancer Discovery, 2021, 2544-2563
|

|
| S1171 |
CYC116
|
CYC116是一种有效的Aurora A/B抑制剂,Ki为8.0 nM/9.2 nM,对VEGFR2(Ki为44 nM)作用稍弱,比作用于CDKs效果强50倍,对PKA, Akt/PKB, PKC没有活性,对GSK-3α/β, CK2, Plk1和SAPK2A.没有抑制效果。Phase 1。 |
- Scientific Reports, April 2, 2024, 7739
- Proceedings of the National Academy of Sciences, April 12, 2022, e2122512119
- Scientific Reports, 2024, 7739
|

|
| S5793 |
Motesanib (AMG-706)
|
Motesanib (AMG-706)是一种具有口服生物活性的受体酪氨酸激酶 (receptor tyrosine kinase)抑制剂,对VEGFR1, VEGFR2, VEGFR3, Kit, PDGFR和Ret的IC50值分别为2、3、6、8、84和59 nM。 |
- Signal Transduct Target Ther, 2022, 7(1):147
- Front Cell Dev Biol, 2022, 10:836179
- J Pharm Sci, 2022, 111(8):2180-2190
|
|
| S8573 |
Sitravatinib (MGCD516)
|
Sitravatinib (MGCD516, MG-516)是一种新型的、靶向多种参与调节S180肉瘤细胞生长的RTKs的小分子抑制剂,包括c-Kit, PDGFRβ, PDGFRα, c-Met和Axl。 |
- Translational Cancer Research, October 2019, 2425-2438
- Cancer Research, October 15, 2025, 3983-3998
- Neuro-Oncology, May 2025, 1372–1384
|
|
| S1220 |
OSI-930
|
OSI-930是一种有效的Kit (c-Kit), KDR和CSF-1R抑制剂,IC50分别为80 nM, 9 nM和15 nM,对Flt-1, c-Raf和Lck具有适度的抑制活性,对PDGFRα/β, Flt-3和Abl抑制活性较弱。Phase 1。 |
- Nat Commun, 2024, 15(1):4739
- Cell Rep, 2019, 28(9):2331-2344
- Nat Biomed Eng, 2018, 2(8):578-588
|

|
| S2859 |
Golvatinib (E7050)
|
Golvatinib (E7050)是c-Met和VEGFR-2双重抑制剂,IC50分别为14 nM和16 nM,不抑制bFGF刺激的HUVEC生长(浓度高达1000 nM)。Phase 1/2。 |
- Int J Mol Sci, 2023, 24(11)9606
- International Journal of Molecular Sciences, 2023, 9606
- International Journal of Molecular Sciences, 2022, 14884
|

|
| S2622 |
PP121
|
PP121是一种作用于PDGFR, Hck, mTOR, VEGFR2, Src和Abl的多靶点抑制剂,IC50分别为2 nM, 8 nM, 10 nM, 12 nM, 14 nM和18 nM,也抑制DNA-PK,IC50为60 nM。 |
- PLOS One, October 21, 2016, e0164895
- Cancer Research, November 01, 2015, 4548-4559
- Life Sci Alliance, 2021, 4(2)e202000882
|

|
| S2366 |
Taxifolin (Dihydroquercetin)
|
Taxifolin(Dihydroquercetin)是一种二氢黄酮醇类,是一种黄酮类化合物。是I型VEGFR2 kinase的抑制剂。 |
- J Virol, 2025, e0209524.
- Foods, 2025, 14(24)4314
- Foods, 2025, 4314
|
|
| S1557 |
KRN 633
|
KRN 633是一种ATP竞争性的VEGFR1/2/3抑制剂,IC50为170 nM/160 nM/125 nM,微弱抑制PDGFR-α/β和c-Kit,对细胞中FGFR-1, EGFR和c-Met的磷酸化没有抑制作用。 |
- Aust Dent J, 2020, 10.1111/adj.12752
- J Med Invest, 2017, 64(3.4):262-265
- Oncotarget, 2016, 7(5):5985-99
|

|
| S5667 |
Fruquintinib (HMPL-013)
|
Fruquintinib is a small molecule inhibitor with strong potency and high selectivity against VEGFR family. It inhibits VEGFR 1, 2, 3, with IC50 values of 33 nM, 35 nM and 0.5 nM, respectively and shows only weak inhibition of RET, FGFR-1 and c-kit kinases. |
- Sci Adv, 2025, 11(20):eads1631
- EMBO J, 2024, 43(8):1519-1544
- iScience, 2024, 27(10):110862
|
|
| S2231 |
Telatinib
|
Telatinib (BAY 57-9352) 是一种有效的VEGFR2/3, c-Kit和PDGFRα抑制剂,IC50分别为6 nM/4 nM, 1 nM和15 nM。Phase 2。 |
- Sci Adv, 2022, 8(6):eabg9455
- Leiden University Scholarly Publications, 2020, N/A
- Cell Stem Cell, 2019, 24(4):654-669
|

|
| S8721 |
PDGFR inhibitor 1
|
PDGFR inhibitor 1是一种具有口服活性的Kit (c-Kit)和PDGFR抑制剂,具有潜在的抗肿瘤活性。它还能抑制好几种其他激酶,包括VEGFR2、TIE2、PDGFR-beta和CSF1R,因此进而抑制肿瘤细胞的生长。 |
- Res Sq, 2025, rs.3.rs-7032881
- bioRxiv, 2025, 2025.06.11.659120
- eLife, 2023, e79840
|
|
| S7258 |
SKLB1002
|
SKLB1002 是一种与ATP竞争的强效VEGFR2抑制剂,其IC50为32 nM. |
- Sci Rep, 2025, 15(1):8177
- Sci Adv, 2024, 10(20):eadl0633
- Dis Model Mech, 2023, 16(5)dmm049571
|

|
| S2201 |
BMS-794833
|
BMS-794833是一种有效的,ATP竞争性Met (c-Met)/VEGFR2抑制剂,IC50为1.7 nM/15 nM,也抑制Ron, Axl和Flt3,IC50低于3 nM;是BMS-817378的前体药物。 Phase 1。 |
- Frontiers in Oncology, August 20, 2021, 686765
- Cell Stem Cell, 2025, 32(11):1671-1690.e13
- Cancer Res, 2021, canres.1428.2021
|
|
| S8189 |
BAW2881 (NVP-BAW2881)
|
BAW2881 (NVP-BAW2881)是一种新型的VEGFR酪氨酸激酶抑制剂。在1.0-4.3 nM浓度下能有效地抑制VEGFR1-3;在45-72 nM的浓度下,抑制PDGFRβ, c-Kit和RET (c-RET)。 |
- J Transl Med, 2025, 23(1):985
- mediaTUM, 2022, 158
- JCI Insight, 2021, 143285
|
|
| S5272 |
Toceranib phosphate
|
Toceranib phosphate (Palladia, SU11654), the phosphate salt of toceranib, is a selective inhibitor of the tyrosine kinase activity of several members of the split kinase RTK family, including Flk-1/KDR, PDGFR, and Kit with Ki values of 6 nM and 5 nM for Flk-1/KDR and PDGFRβ, respectively. |
- Frontiers in Veterinary Science, May 21, 2024, 1392728
- Journal of Pharmacology and Experimental Therapeutics, February 15, 2024, 774-787
- Liver International, April 2023, 928-944
|
|
| S7003 |
AZD2932
|
AZD2932 是一种有效的多靶点蛋白酪氨酸激酶抑制剂,对VEGFR-2,PDGFRβ,Flt-3,和 c-Kit 的 IC50 分别为 8 nM,4 nM,7 nM,和 9 nM。
|
- Cancer Research, 2021, 747-762
- Cancer Res, 2020, canres.1992.2020
- Molecules, 2020, 8;25(9) pii: E2220
|
|
| S0504 |
SU14813
|
SU14813 (SU 014813)是一种多受体 tyrosine kinase 的抑制剂,对于 VEGFR2、VEGFR1、PDGFRβ 和 Kit (c-Kit) 的IC50值分别为50 nM,2 nM,4 nM和15 nM。SU14813 具有强大的抗血管生成和抗肿瘤活性。 |
- Cancer Cell, 2022, S1535-6108(22)00312-9
- Adv Sci (Weinh), 2021, e2101848
- Advanced Science, 2021, e2101848
|
|
| S0278 |
SU5614
|
SU5614 (Chloro-SU5416, Chloro-Semaxanib) 是一种小分子 receptor tyrosine kinases (RTK) 的抑制剂,对 VEGFR-2、c-kit 以及野生型和突变型 FLT3 均有抑制作用。SU5614 可减少细胞增殖并诱导凋亡。 |
- Frontiers in Cell and Developmental Biology, January 26, 2022, 797047
- Advanced Science, August 7, 2021, e2101848
- Am J Pathol, 2024, S0002-9440(24)00326-2
|
|
| S0765 |
MAZ51
|
MAZ51 是一种选择性的 vascular endothelial growth factor receptor (VEGFR)-3 (Flt-4) tyrosine kinase 的有效抑制剂。MAZ51 通过 Akt/GSK3β 的磷酸化和 RhoA 的激活来诱导神经胶质瘤细胞的细胞圆缩和G2/M细胞周期停滞。 MAZ51 可抑制多种非VEGFR-3表达肿瘤细胞的增殖并诱导其凋亡。 |
- Sci Rep, 2025, 15(1):1283
- International Immunopharmacology, 2025, 114443
- Human Molecular Genetics, 2014, 6553-6566
|
|
| S6412 |
Altiratinib
|
Altiratinib (DCC-2701) 是有效的TRK, Met (c-Met), TIE2和VEGFR2激酶抑制剂,对TRKA, B和C的IC50分别为0.9 nM, 4.6 nM和0.8 nM。它抑制Met (c-Met)和Met (c-Met)突变体的IC50范围为0.3-6 nM。 |
- J Microbiol, 2025, 63(2):e2409001
- Theranostics, 2021, 11(20):9918-9936
- Cold Spring Harb Mol Case Stud, 2021, 7(5)a006109
|
|
| S1138 |
Brivanib Alaninate (BMS-582664)
|
Brivanib Alaninate (BMS-582664)是BMS-540215的前体药物,是一种ATP竞争性的VEGFR2抑制剂,IC50为25 nM。 |
- Int J Oncol, 2014, 44(3):959-69
- Assay Drug Dev Technol, 2014, 12(9-10):514-26
|
|
| S2018 |
ENMD-2076 L-(+)-Tartaric acid
|
ENMD-2076 L-(+)-Tartaric acid 是ENMD-2076的酒石酸,选择性作用于Aurora A和VEGFR(Flt3),IC50分别为14 nM和1.86 nM,作用于Aurora A比作用于Aurora B选择性高25倍,对VEGFR2/KDR和VEGFR3, FGFR1和FGFR2和PDGFRα作用效果稍弱。Phase 2。 |
- Veterinary and Comparative Oncology, 2016 Mar, 14(1):13-27
- International Journal of Oncology, 2015 Jul, 47(1):122-32
|
|
| S6535 |
SU1498
|
SU1498是KDR抑制剂,IC50为0.7 μM,可在内皮细胞中刺激磷酸化ERK1/2的累积。 |
- Cell Metab, 2020, 5;31(5):892-908 e11
- Biol Open, 2018,
|
|
| S7688 |
Ki20227
|
Ki20227 是一种口服活性的 c-Fms tyrosine kinase(CSF1R) 的高选择性抑制剂,对 c-Fms、vascular endothelial growth factor receptor-2 (KDR/VEGFR-2)、stem cell factor receptor (c-Kit) 和 platelet-derived growth factor receptor beta (PDGFRβ) 的IC50值为2 nM、12 nM、451 nM和217 nM。 |
- Cancer Immunol Immunother, 2024, 73(5):76
- Arteriosclerosis, Thrombosis, and Vascular Biology, 2023, 1063-1078
|
|
| S6526 |
SKLB 610
|
SKLB-610是一种多靶点的酪氨酸激酶 (tyrosine kinases)抑制剂。它对VEGFR2的抑制作用最为有效,是FGFR2和PDGFR的弱抑制剂。 |
- Biomed Pharmacother, 2022, 149:112922
- Biomedicine & Pharmacotherapy, 2022, 112922
- International Journal of Molecular Sciences, 2022, 7853
|
|
| S6514 |
SU5408
|
SU5408 (VEGFR2 Kinase Inhibitor I)是一种有效、选择性 VEGFR2 Kinase抑制剂,IC50为70 nM。 |
- Transl Oncol, 2022, 25:101516
- Translational Oncology, 2022, 101516
- J Dent Sci, 2022, 17(4):1471-1479
|
|
| S0487 |
Sulfatinib
|
Sulfatinib是一种有效的、具有高度选择性的 tyrosine kinase 的抑制剂,对于 VEGFR1、VEGFR2、VEGFR3、FGFR1 和 CSF1R 的IC50值分别为2 nM、24 nM、1 nM、15 nM和4 nM。Sulfatinib 在针对晚期NET患者的实验中显示出令人鼓舞的抗肿瘤活性和可控毒性。 |
- International Journal of Biological Sciences, 2025, 7043-7062
- Frontiers in Oncology, 2023, 1158857
- Front Oncol, 2023, 13:1158857
|
|
| S0290 |
SU5204
|
SU5204 是一种酪氨酸激酶抑制剂,对 FLK-1 (VEGFR-2) 和 HER2 的IC50值分别为4 μM和51.5 μM。 |
- Front Pharmacol, 2021, 12:804327
|
|
| S2211 |
AG-13958
|
AG-13958 (AG-013958)是VEGFR酪氨酸激酶抑制剂,在临床开发中发现ST给药可治疗与年龄相关性黄斑变性(AMD)的脉络膜新生血管生成。 |
- iScience, 2023, 107548
|
|
| S7647 |
Lucitanib (E3810) hydrochloride
|
Lucitanib (E-3810, AL3810) hydrochloride 是 Vascular endothelial growth factor receptor (VEGFR) 和 Fibroblast growth factor receptor (FGFR) 的双重抑制剂。 Lucitanib hydrochloride (E-3810, AL3810) 有效且选择性地抑制 VEGFR1、VEGFR2、VEGFR3、FGFR1 和 FGFR2,IC50值分别为7 nM、25 nM、10 nM、17.5 nM和82.5 nM。 |
- Sci Rep, 2023, 13(1):20223
|
|
| S6520 |
WHI-P180
|
WHI-P180是多种激酶的抑制剂,对RET (c-RET)和KDR的IC50值分别为4.5 nM和66 nM。 |
|
|
| S6543 |
ZD-4190
|
ZD-4190是VEGF RTK抑制剂,对KDR和Flt-1的IC50值分别为29 ± 4 nM和708 ± 63 nM。 |
|
|
| S8188 |
BFH772
|
BFH772是一种新型的、有效的、可口服的VEGFR2抑制剂,IC50为3 nM。 |
|
|
| S0291 |
SU5208
|
SU5208(3-[(Thien-2-yl)methylene]-2-indolinone)是血管内皮生长因子受体2(vascular endothelial growth factor receptor-2, VEGFR2)的抑制剂。 |
|
|
| E2397 |
hVEGF-IN-1
|
hVEGF-IN-1是一种喹唑啉衍生物,可特异性结合到核糖体内部进入位点A(internal ribosome entry site A, IRES-A)的G-rich序列,Kd为0.928 μM,从而破坏G -四复合物结构;并可降低VEGF-A蛋白表达,从而阻碍肿瘤细胞迁移,抑制肿瘤生长。 |
|
|
| S8882 |
ODM-203
|
ODM-203是FGFR 和 VEGFR的双重抑制剂,对重组FGFR1,FGFR2,FGFR3,FGFR4,VEGFR1,VEGFR2和VEGFR3的ic50分别为11 nM,16 nM,6 nM,35 nM,26 nM,9 nM,5 nM。 |
|
|
| S0763 |
Tyrphostin AG1433
|
Tyrphostin AG1433 (AG1433, SU1433)是一种选择性血小板衍生生长因子受体β (platelet-derived growth factor receptor β, PDGFRβ)和血管内皮生长因子受体2 (vascular endothelial growth factor receptor 2, VEGFR-2, Flk-1/KDR) 抑制剂,IC50分别为5.0 μM和9.3 μM。 |
|
|
| E0499 |
4SC-203
|
4SC-203(SC71710)是一种具有潜在抗肿瘤活性的多激酶抑制剂。4SC-203选择性抑制FMS相关酪氨酸激酶3(FMS-related tyrosine kinase 3, FLT3/STK1)、FLT3的突变形式和血管内皮生长因子受体(vascular endothelial growth factor receptors, VEGFRs)。
|
|
|
| E2656 |
R1530
|
R1530是一种有效的、口服活性的、双重作用的有丝分裂/血管生成抑制剂,可抑制血管内皮生长因子受体2 (vascular endothelial growth factor receptor 2, VEGFR2)和成纤维细胞生长因子受体1(fibroblast growth factor receptors 1, FGFR1),IC50分别为10 nM和28 nM。 |
|
|
| S9621 |
Donafenib (Sorafenib D3)
|
Donafenib (Sorafenib D3, Bay 43-9006 D3, CM-4307)是氘标记的Sorafenib。Sorafenib 是一种多激酶的抑制剂,对 Raf-1、mVEGFR-2、mVEGFR-3 和 B-RAF 的抑制作用对应的IC50值分别为6 nM、15 nM、20 nM和22 nM。 |
|
|
| E1078 |
Emvododstat (PTC299)
|
Emvododstat(PTC299)是致病性VEGF和双氢乳清酸酯脱氢酶(dihydroorotate dehydrogenase, DHODH)的转录后抑制剂,可抑制VEGFAmRNA翻译和异常细胞增殖。在HeLa细胞中,PTC299抑制缺氧诱导的VEGFA蛋白的产生,EC50为1.64 nM。 |
|
|
| S5607 |
4,4'-Bis(4-aminophenoxy)biphenyl
|
4,4'-Bis(4-aminophenoxy)biphenyl is a monomer for polyimide production. |
|
|
| E0616 |
Chiauranib
|
Chiauranib (CS2164)能够选择性抑制多种激酶靶标,包括aurora B激酶(aurora B kinase, AURKB)、集落刺激因子1受体(colony-stimulating factor 1 receptor, CSF1R)和血管内皮生长因子受体(vascular endothelial growth factor receptor, VEGFR)/血小板衍生生长因子受体(platelet-derived growth factor receptor, PDGFR)/c -Kit,从而抑制肿瘤细胞的快速增殖,增强抗肿瘤免疫,抑制肿瘤血管生成,达到抗肿瘤功效。 |
|
|
| S6870 |
Ningetinib
|
Ningetinib (CT-053, DE-120, CT053PTSA)是一种有效的、可口服的 tyrosine kinase,对 c-Met、VEGFR2 和 Axl 的IC50值分别为6.7 nM、1.9 nM和 <1.0 nM。Ningetinib 表现出了抗肿瘤的活性。 |
|
|
| S6843 |
Vorolanib
|
Vorolanib (X-82,CM082)是一种口服的 vascular endothelial growth factor receptor (VEGFR) 和 platelet-derived growth factor receptor (PDGFR) 的多激酶双重抑制剂,并具有抗血管生成和抗肿瘤的活性。 |
|
|
| S6764 |
Pamufetinib (TAS-115)
|
Pamufetinib (TAS-115) 是一种高效的 c-Met 和 VEGFR 双重抑制剂,对重组 VEGFR2 和重组 MET 的 IC50 分别为 30 nM 和 32 nM。 |
|
|
| S6939 |
Nastorazepide
|
Nastorazepide (Z-360)是一种胆囊收缩素-2/胃泌素受体(cholecystokinin-2/gastrin receptor, CCK2/胃泌素受体)拮抗剂,与吉西他滨联合使用可延长胰腺癌原位异种移植小鼠的存活率并降低吉西他滨诱导的血管内皮生长因子(vascular endothelial growth factor, VEGF)表达。 |
|
|
| S0377 |
CS-2660 (JNJ-38158471)
|
CS-2660 (JNJ-38158471) 是一种耐受性良好的、口服有效的、高选择性的 VEGFR-2 抑制剂,IC50值为40 nM。CS-2660(JNJ-38158471)还抑制紧密相关的酪氨酸激酶,如 Ret (c-RET) 和 Kit (c-Kit),IC50为180 nM和500 nM,但无明显活性 (>1 microM) 针对VEGFR-1和VEGFR-3。 |
|
|
| E0534 |
WAY-340935
|
WAY-340935 (VEGFR2-IN-2)可以抑制VEGFR2的功能,其对H460细胞系的抗增殖活性部分是通过与VEGFR蛋白的相互作用而产生的。 |
|
|
| E3414 |
Semen litchi Extract
|
Semen litchi Extract是从荔枝Litchi chinensis中提取的,具有抗肝癌作用,并通过靶向VEGFR抑制肝细胞癌血管生成。 |
|
|
| E4914 |
Cabozantinib hydrochloride
|
Cabozantinib hydrochloride(XL184, BMS-907351 hydrochloride) 是一种有效的 c-MET 和 VEGFR2 小分子激酶抑制剂,IC50 分别为 1.3 nM、0.035 nM。它还抑制 RET、KIT、AXL、Tie2 和 FLT3,IC50 分别为 5.2 nM、4.6 nM、7 nM、14.3 nM、11.3 nM。它可以成为抑制 MET 和 VEGFR 信号失调的癌症中的肿瘤血管生成和转移的有希望的药物。 |
|
|
| E0207 |
Chebulinic acid
|
Chebulinic acid,一种从榄仁树果实中分离的酚类化合物,是一种新型流感病毒神经氨酸酶(Influenza viral neuraminidase)抑制剂。Chebulinic acid 也是一种抗血管生成剂,通过抑制 VEGF 起作用。Chebulinic acid 具有抗肿瘤活性。 |
|
|
| E3774 |
Chlorella Pyrenoidosa Extract
|
Chlorella Pyrenoidosa Extract是从小球藻Chlorella Pyrenoidosa中提取的,对VEGF诱导的新生血管有明显的抗血管生成活性和抗增殖活性。 |
|
|
| E4694 |
EVT801
|
EVT801 是一种强效且选择性的 VEGFR-3 抑制剂,IC50 为 11 nM。它具有强效的抗肿瘤作用,是一种有前途的抗淋巴管生成药物,适用于 VEGFR-3 阳性肿瘤患者。 |
|
|
| S6808 |
SU5205
|
SU5205是 VEGF receptor 2 (VEGFR2/FLK-1) 的抑制剂,IC50为9.6 µM。 |
|
|
| S6809 |
SU5214
|
SU5214是 VEGF receptor 2 (VEGFR2/FLK-1) 和 EGFR 的抑制剂,对应的IC50值分别为14.8 µM 和 36.7 µM。 |
|
|
| E1435 |
Tinengotinib
|
Tinengotinib (TT-00420) 是一种新型多激酶抑制剂,可强力抑制 Aurora A/B,IC50 值分别为 1.2/3.3 nM。 它还对 FGFR1/2/3、VEGFR1、JAK1/2 和 CSF1R 表现出有效的抑制活性,可用于治疗三阴性乳腺癌 (TNBC) 。 |
|
|
| E0142 |
XL092
|
XL092 (JUN04542) 是多种 RTK 的 ATP 竞争性抑制剂,包括 MET、VEGFR2、AXL 和 MER,在基于细胞的测定中 IC50 值分别为 15 nM、1.6 nM、3.4 nM 和 7.2 nM。 |
|
|











































